

Rapid evolution at the Drosophila telomere: transposable element dynamics at an intrinsically unstable locus
- PMID: 33724410
- PMCID: PMC8045721
- DOI: 10.1093/genetics/iyaa027
Rapid evolution at the Drosophila telomere: transposable element dynamics at an intrinsically unstable locus
Abstract
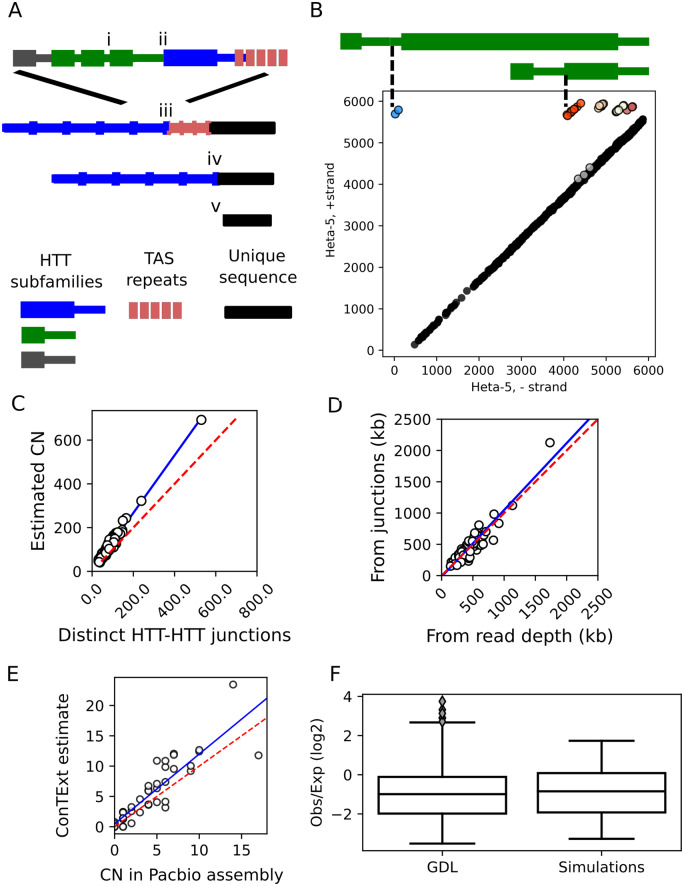
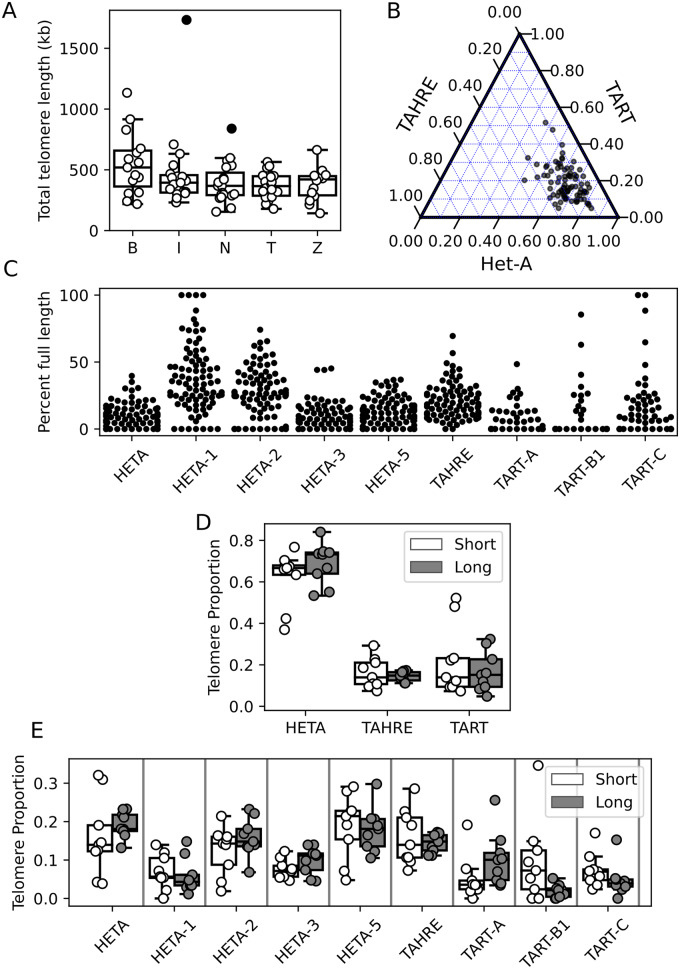
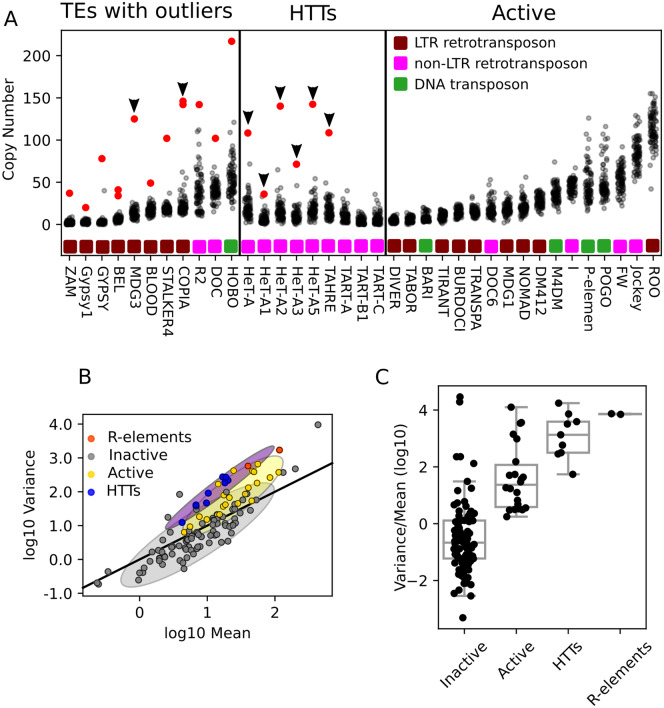
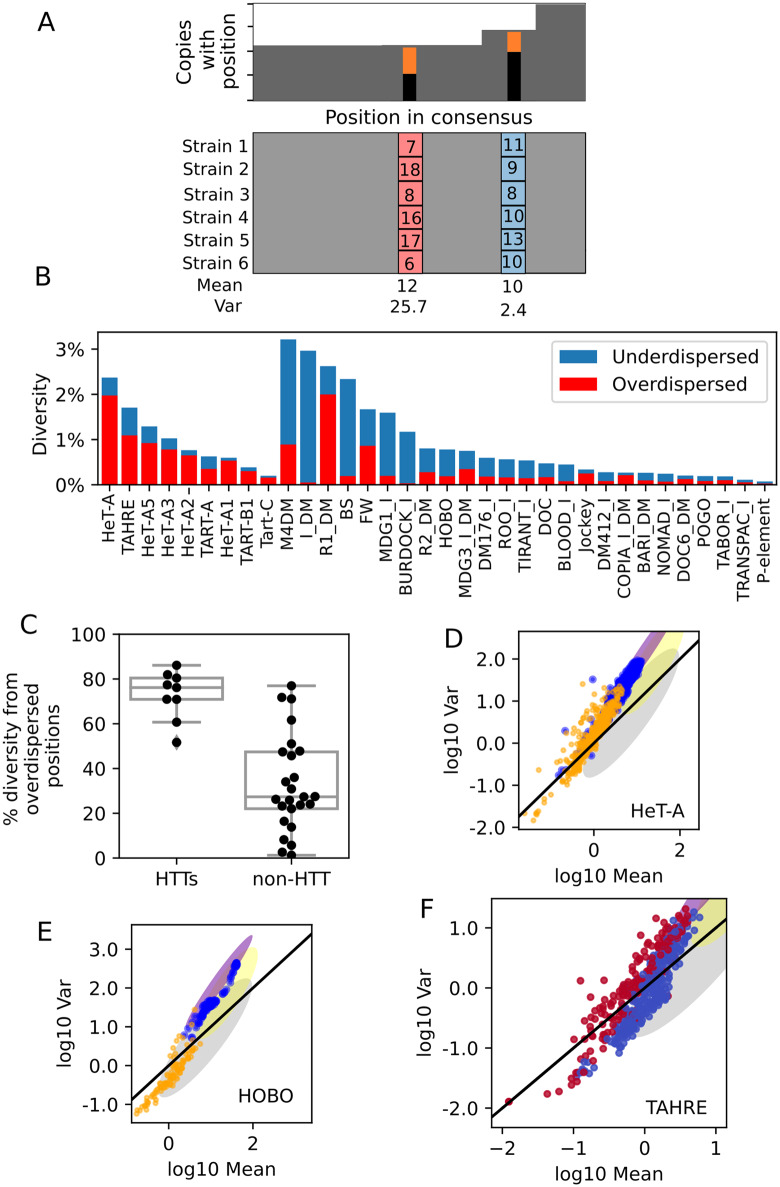
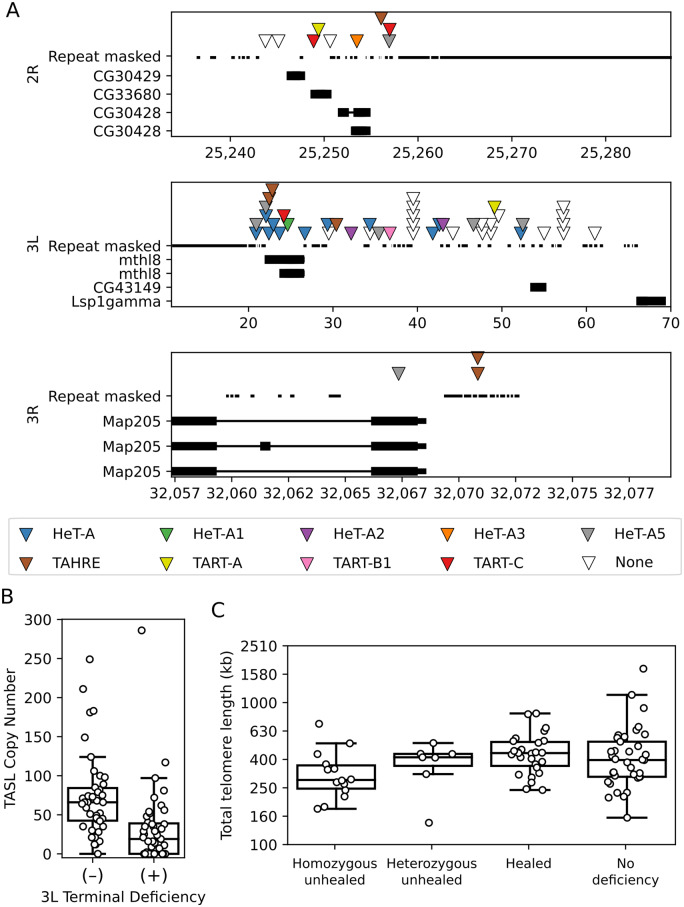
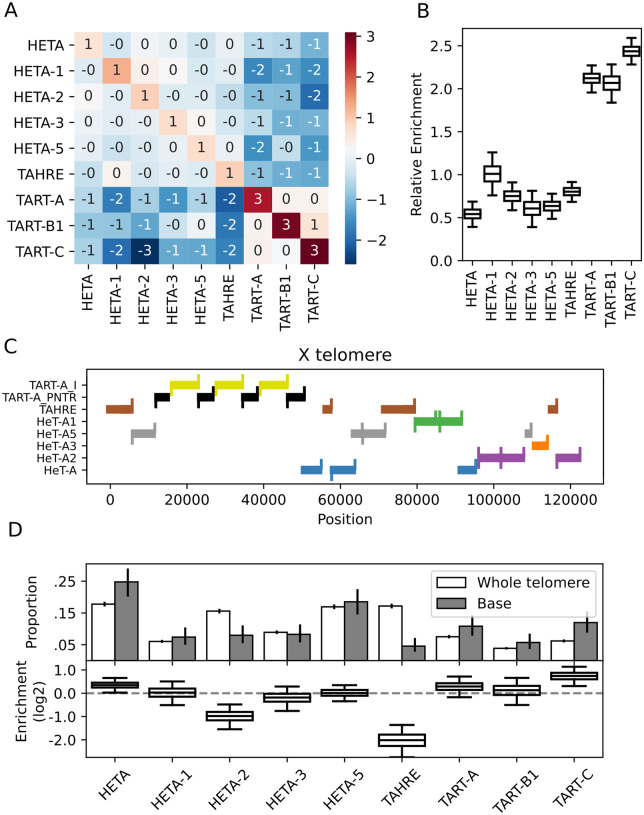
Drosophila telomeres have been maintained by three families of active transposable elements (TEs), HeT-A, TAHRE, and TART, collectively referred to as HTTs, for tens of millions of years, which contrasts with an unusually high degree of HTT interspecific variation. While the impacts of conflict and domestication are often invoked to explain HTT variation, the telomeres are unstable structures such that neutral mutational processes and evolutionary tradeoffs may also drive HTT evolution. We leveraged population genomic data to analyze nearly 10,000 HTT insertions in 85 Drosophila melanogaster genomes and compared their variation to other more typical TE families. We observe that occasional large-scale copy number expansions of both HTTs and other TE families occur, highlighting that the HTTs are, like their feral cousins, typically repressed but primed to take over given the opportunity. However, large expansions of HTTs are not caused by the runaway activity of any particular HTT subfamilies or even associated with telomere-specific TE activity, as might be expected if HTTs are in strong genetic conflict with their hosts. Rather than conflict, we instead suggest that distinctive aspects of HTT copy number variation and sequence diversity largely reflect telomere instability, with HTT insertions being lost at much higher rates than other TEs elsewhere in the genome. We extend previous observations that telomere deletions occur at a high rate, and surprisingly discover that more than one-third do not appear to have been healed with an HTT insertion. We also report that some HTT families may be preferentially activated by the erosion of whole telomeres, implying the existence of HTT-specific host control mechanisms. We further suggest that the persistent telomere localization of HTTs may reflect a highly successful evolutionary strategy that trades away a stable insertion site in order to have reduced impact on the host genome. We propose that HTT evolution is driven by multiple processes, with niche specialization and telomere instability being previously underappreciated and likely predominant.
Keywords: Drosophila; genomic conflict; telomere; terminal deletions; transposable element.
© The Author(s) 2020. Published by Oxford University Press on behalf of Genetics Society of America. All rights reserved. For permissions, please email: journals.permissions@oup.com.
Figures
Similar articles
-
TAHRE, a novel telomeric retrotransposon from Drosophila melanogaster, reveals the origin of Drosophila telomeres.Mol Biol Evol. 2004 Sep;21(9):1620-4. doi: 10.1093/molbev/msh180. Epub 2004 Jun 2. Mol Biol Evol. 2004. PMID: 15175413
-
VHICA, a New Method to Discriminate between Vertical and Horizontal Transposon Transfer: Application to the Mariner Family within Drosophila.Mol Biol Evol. 2016 Apr;33(4):1094-109. doi: 10.1093/molbev/msv341. Epub 2015 Dec 18. Mol Biol Evol. 2016. PMID: 26685176 Free PMC article.
-
Dynamics and Impacts of Transposable Element Proliferation in the Drosophila nasuta Species Group Radiation.Mol Biol Evol. 2022 May 3;39(5):msac080. doi: 10.1093/molbev/msac080. Mol Biol Evol. 2022. PMID: 35485457 Free PMC article.
-
Maintaining Telomeres without Telomerase in Drosophila: Novel Mechanisms and Rapid Evolution to Save a Genus.Cold Spring Harb Perspect Biol. 2025 Mar 3;17(3):a041708. doi: 10.1101/cshperspect.a041708. Cold Spring Harb Perspect Biol. 2025. PMID: 39694814 Review.
-
Drosophila telomeres: an example of co-evolution with transposable elements.Genome Dyn. 2012;7:46-67. doi: 10.1159/000337127. Epub 2012 Jun 25. Genome Dyn. 2012. PMID: 22759813 Review.
Cited by
-
LINE-1 and SINE-B1 mapping and genome diversification in Proechimys species (Rodentia: Echimyidae).Life Sci Alliance. 2022 Mar 18;5(6):e202101104. doi: 10.26508/lsa.202101104. Print 2022 Jun. Life Sci Alliance. 2022. PMID: 35304430 Free PMC article.
-
Functional Diversification of Chromatin on Rapid Evolutionary Timescales.Annu Rev Genet. 2021 Nov 23;55:401-425. doi: 10.1146/annurev-genet-071719-020301. Annu Rev Genet. 2021. PMID: 34813351 Free PMC article. Review.
-
Epigenetic silencing and genome dynamics determine the fate of giant virus endogenizations in Acanthamoeba.BMC Biol. 2025 Jul 1;23(1):171. doi: 10.1186/s12915-025-02280-1. BMC Biol. 2025. PMID: 40597104 Free PMC article.
-
Mod(mdg4) variants repress telomeric retrotransposon HeT-A by blocking subtelomeric enhancers.Nucleic Acids Res. 2022 Nov 11;50(20):11580-11599. doi: 10.1093/nar/gkac1034. Nucleic Acids Res. 2022. PMID: 36373634 Free PMC article.
-
Evolutionary dynamics between transposable elements and their host genomes: mechanisms of suppression and escape.Curr Opin Genet Dev. 2023 Oct;82:102092. doi: 10.1016/j.gde.2023.102092. Epub 2023 Jul 28. Curr Opin Genet Dev. 2023. PMID: 37517354 Free PMC article. Review.
References
-
- Abad JP, De Pablos B, Osoegawa K, De Jong PJ, Martín-Gallardo A, et al. 2004. TAHRE, a novel telomeric retrotransposon from Drosophila melanogaster, reveals the origin of Drosophila telomeres. Mol Biol Evol. 21:1620–1624. - PubMed
-
- Arkhipova IR. 2012. Telomerase, retrotransposons, and evolution. In: Lue NF, Autexier C editors. Telomerases. Hoboken, NJ: John Wiley & Sons, Inc, p. 265–299.
-
- Begun DJ, Aquadro CF. 1995. Evolution at the tip and base of the X chromosome in an African population of Drosophila melanogaster. Mol Biol Evol. 12:382–390. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
