

Astrocytes respond to a neurotoxic Aβ fragment with state-dependent Ca2+ alteration and multiphasic transmitter release
- PMID: 33726852
- PMCID: PMC7968286
- DOI: 10.1186/s40478-021-01146-1
Astrocytes respond to a neurotoxic Aβ fragment with state-dependent Ca2+ alteration and multiphasic transmitter release
Abstract
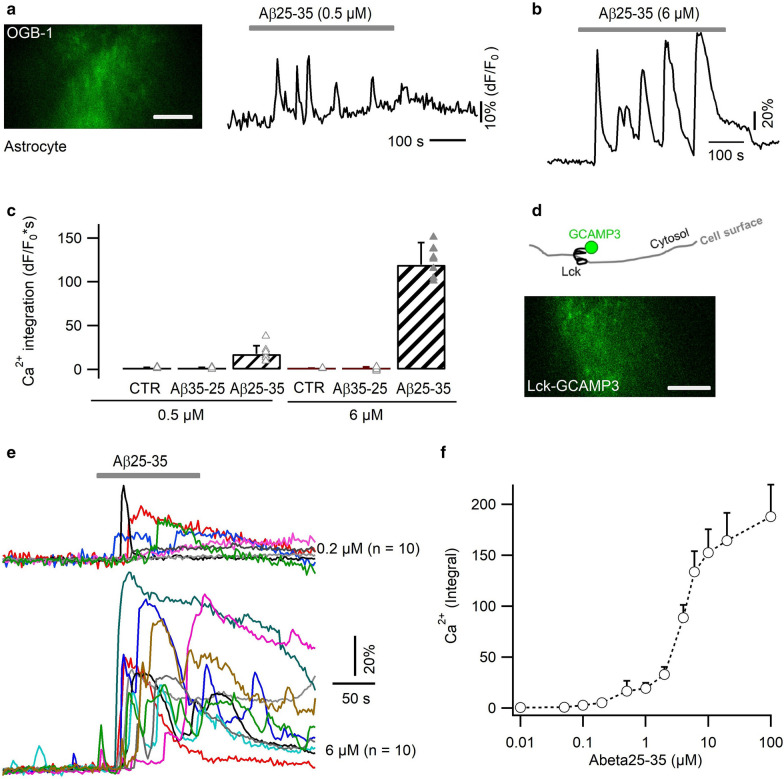
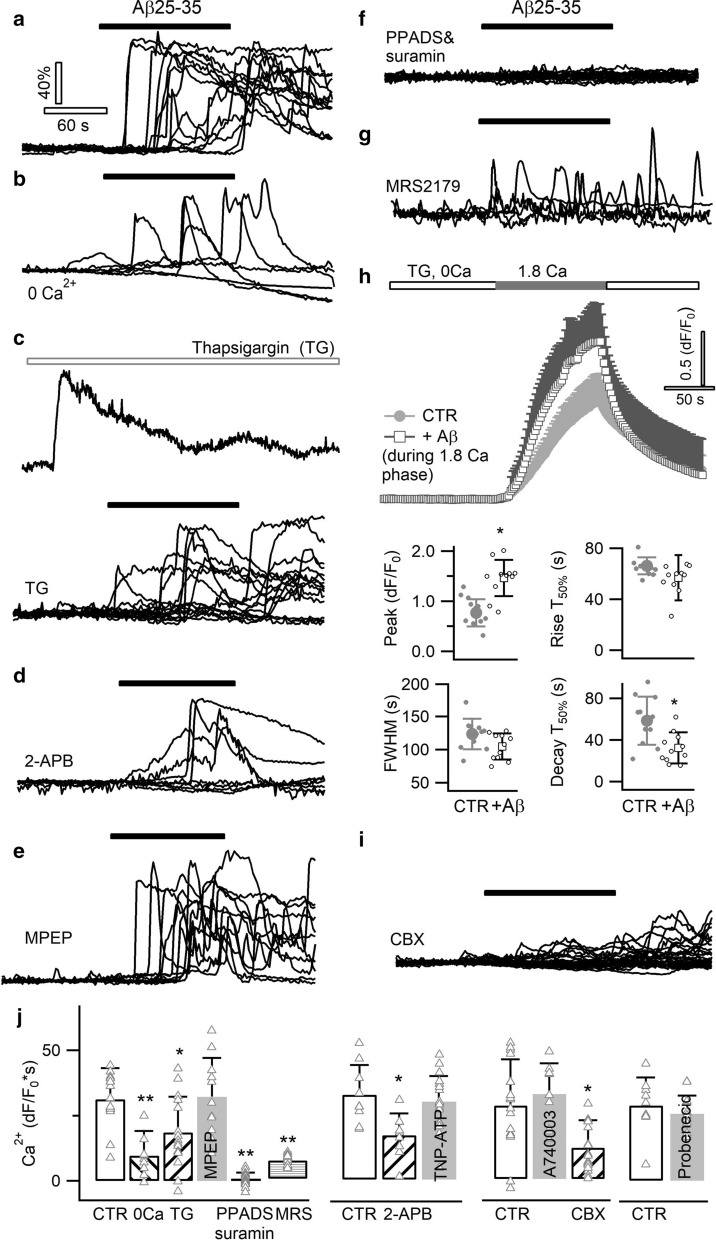
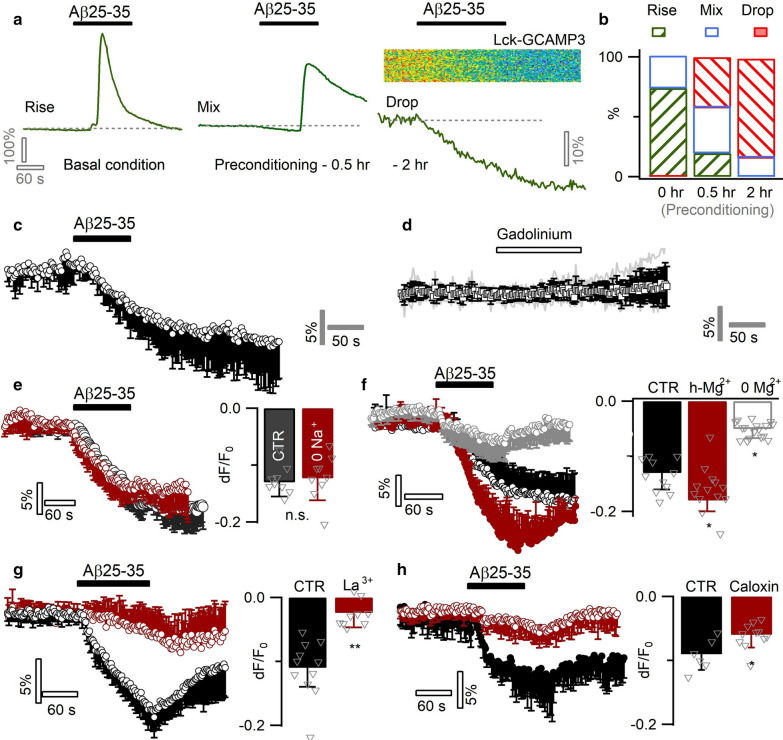
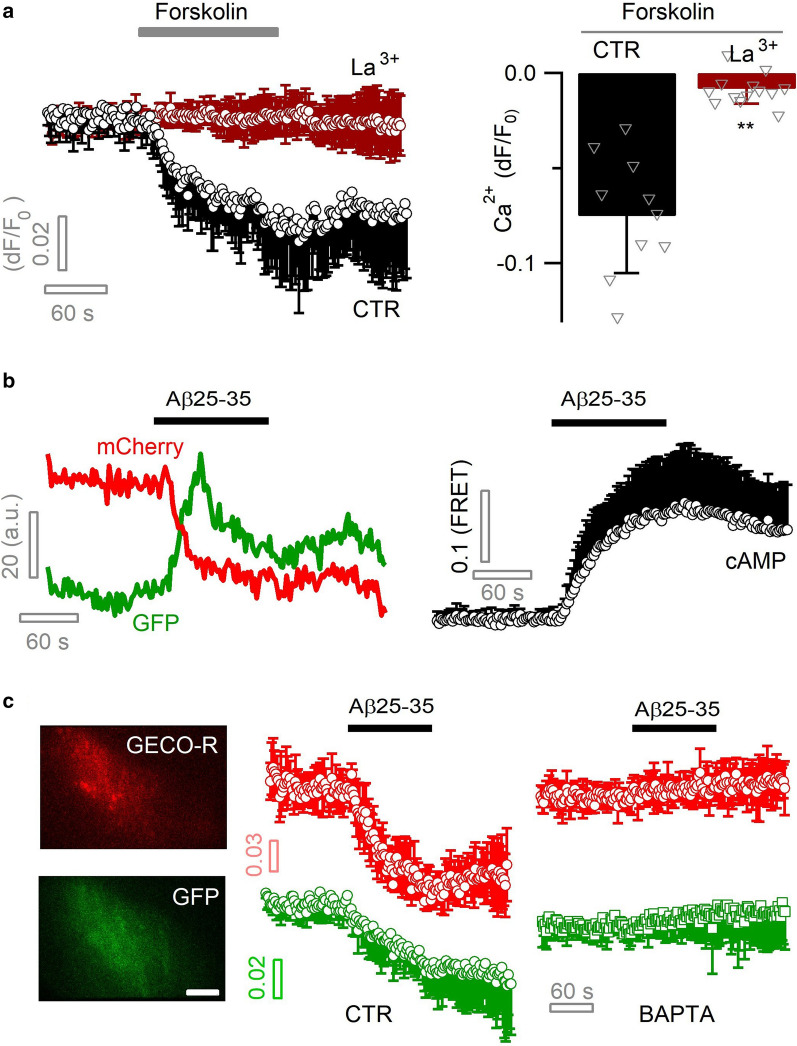
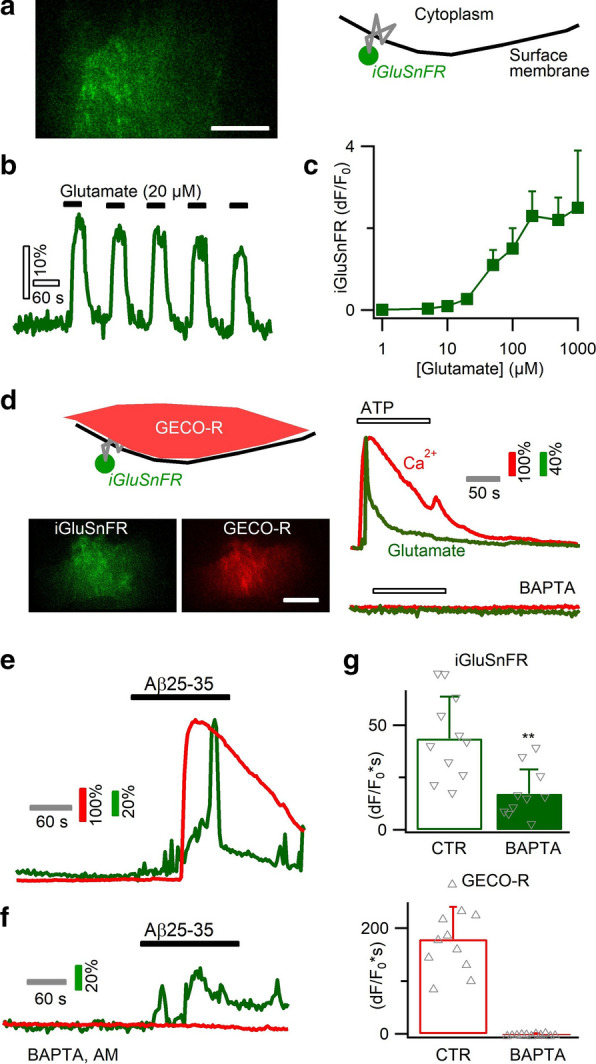
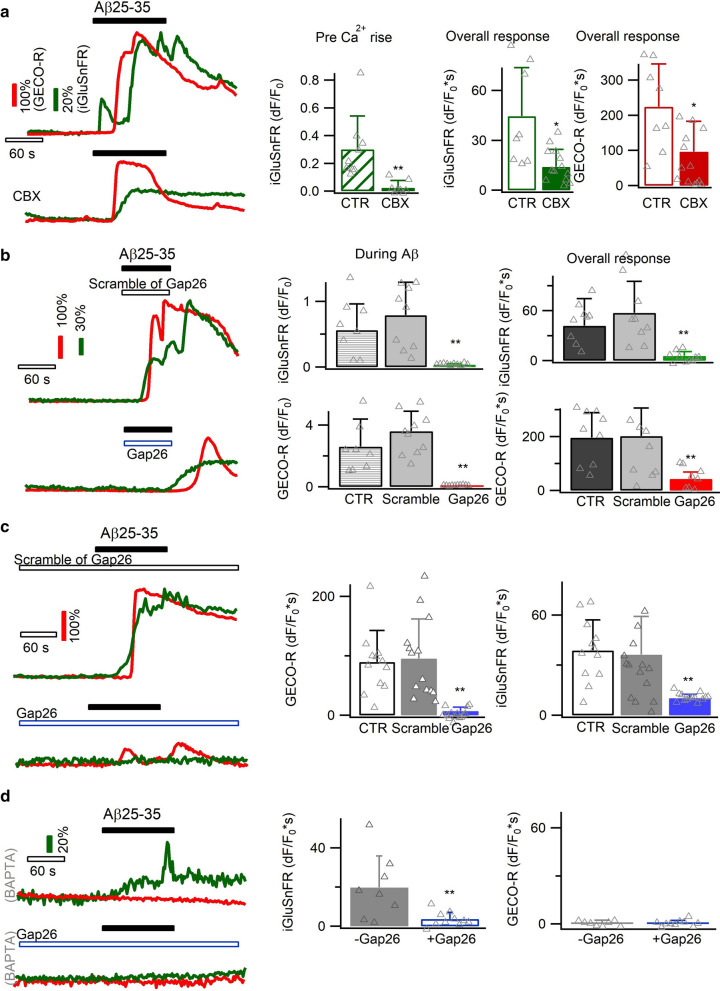
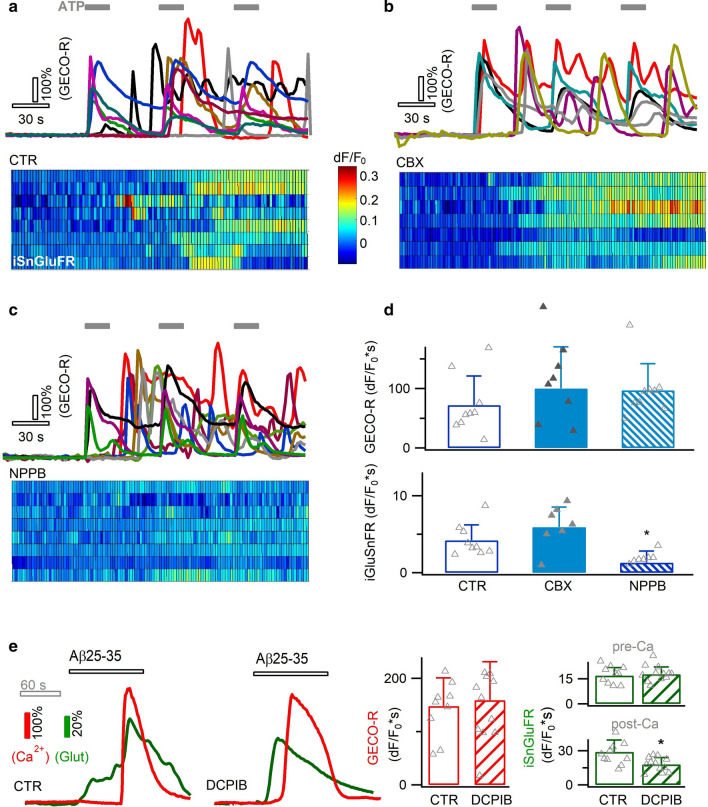
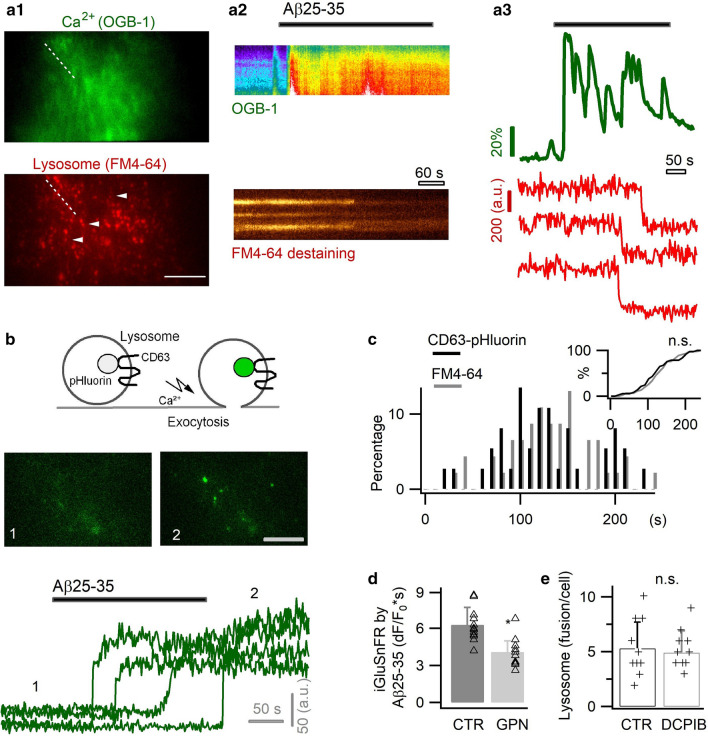
Excessive amounts of amyloid β (Aβ) peptide have been suggested to dysregulate synaptic transmission in Alzheimer's disease (AD). As a major type of glial cell in the mammalian brain, astrocytes regulate neuronal function and undergo activity alterations upon Aβ exposure. Yet the mechanistic steps underlying astrocytic responses to Aβ peptide remain to be elucidated. Here by fluorescence imaging of signaling pathways, we dissected astrocytic responses to Aβ25-35 peptide, a neurotoxic Aβ fragment present in AD patients. In native health astrocytes, Aβ25-35 evoked Ca2+ elevations via purinergic receptors, being also dependent on the opening of connexin (CX) hemichannels. Aβ25-35, however, induced a Ca2+ diminution in Aβ-preconditioned astrocytes as a result of the potentiation of the plasma membrane Ca2+ ATPase (PMCA). The PMCA and CX protein expression was observed with immunostaining in the brain tissue of hAPPJ20 AD mouse model. We also observed both Ca2+-independent and Ca2+-dependent glutamate release upon astrocytic Aβ exposure, with the former mediated by CX hemichannel and the latter by both anion channels and lysosome exocytosis. Our results suggest that Aβ peptide causes state-dependent responses in astrocytes, in association with a multiphasic release of signaling molecules. This study therefore helps to understand astrocyte engagement in AD-related amyloidopathy.
Keywords: ATP; Alzheimer’s disease; Glutamate; Hemichannel; Lysosome.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Alberdi E, Wyssenbach A, Alberdi M, Sanchez-Gomez MV, Cavaliere F, Rodriguez JJ, Verkhratsky A, Matute C. Ca(2+)-dependent endoplasmic reticulum stress correlates with astrogliosis in oligomeric amyloid beta-treated astrocytes and in a model of Alzheimer's disease. Aging Cell. 2013;12:292–302. doi: 10.1111/acel.12054. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
