

Phylo-geo-network and haplogroup analysis of 611 novel coronavirus (SARS-CoV-2) genomes from India
- PMID: 33727249
- PMCID: PMC7994317
- DOI: 10.26508/lsa.202000925
Phylo-geo-network and haplogroup analysis of 611 novel coronavirus (SARS-CoV-2) genomes from India
Abstract
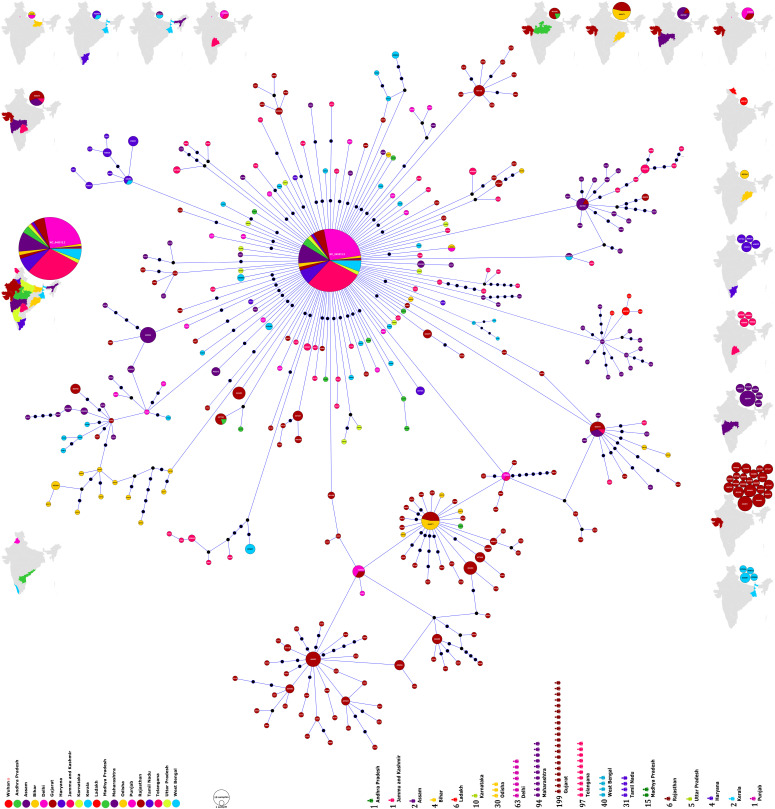
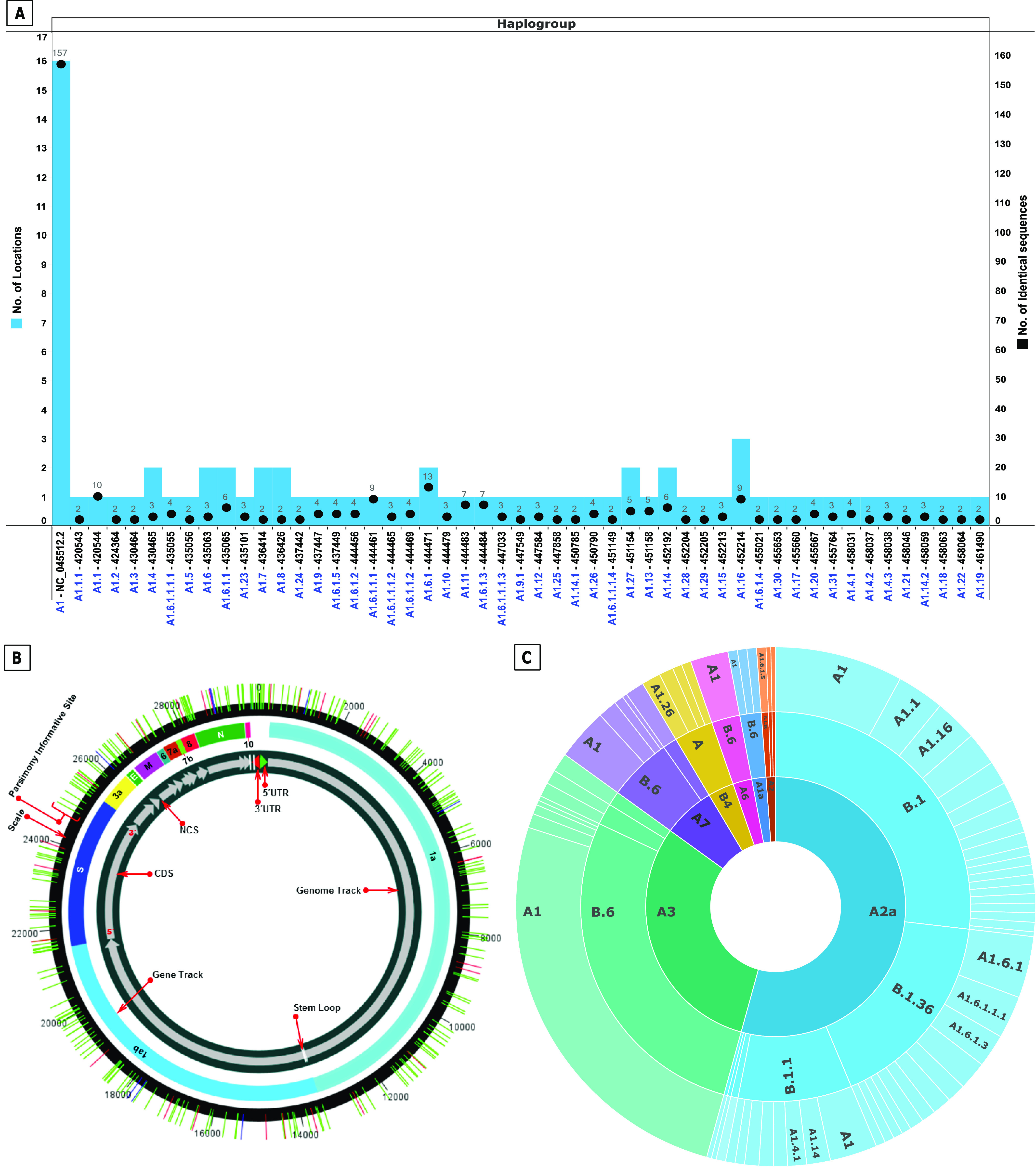
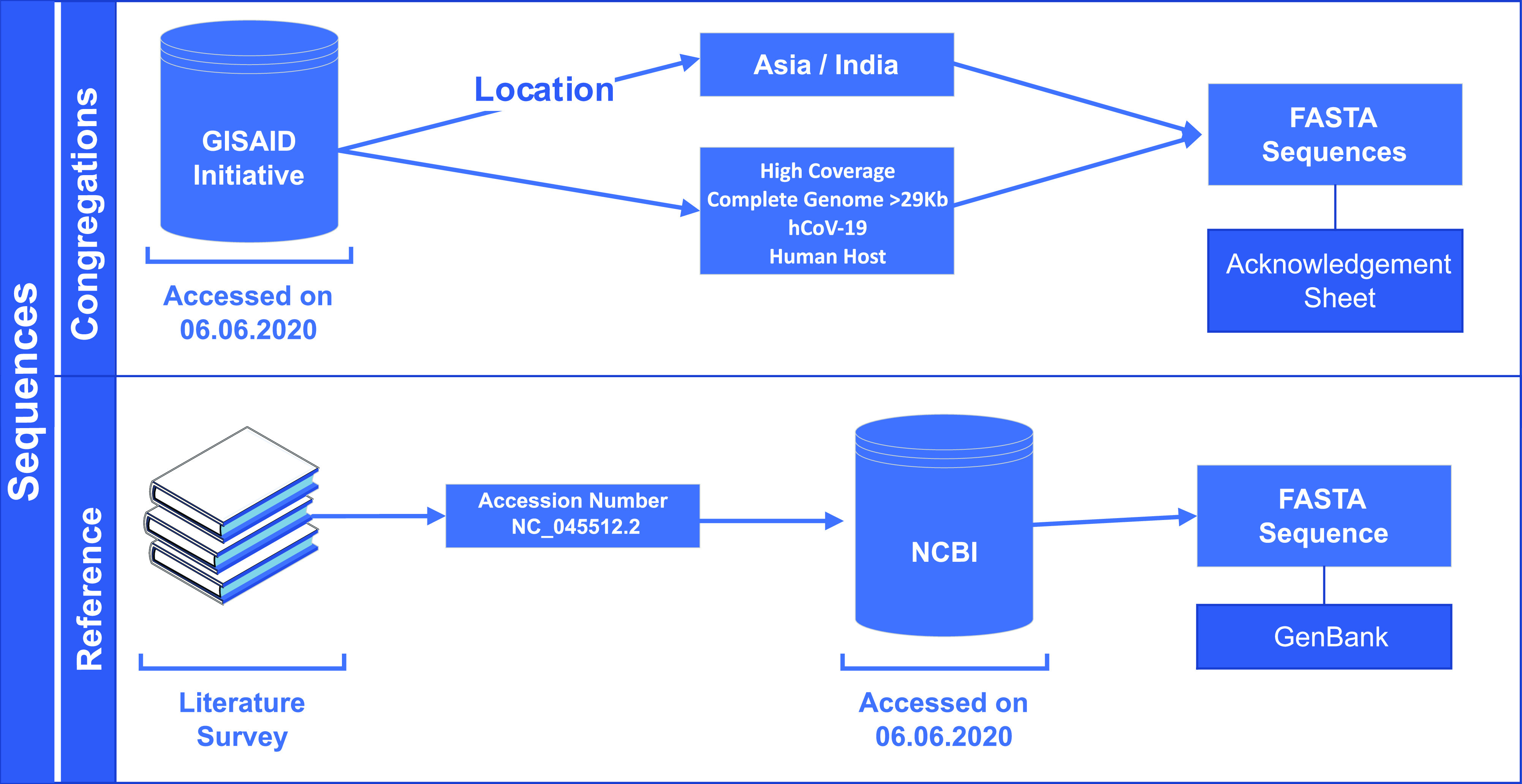
The novel coronavirus (SARS-CoV-2) from Wuhan China discovered in December 2019 has since developed into a global epidemic. Presently, we constructed and analyzed the phylo-geo-network of SARS-CoV-2 genomes from across India to understand the viral evolution in the country. A total of 611 full-length genomes from different states of India were extracted from the EpiCov repository of GISAID initiative on 6 June, 2020. Their alignment with the reference sequence (Wuhan, NCBI accession number NC_045512.2) uncovered 270 parsimony informative sites. Furthermore, 339 genomes were divided into 51 haplogroups. The network revealed the core haplogroup as that of reference sequence NC_045512.2 (Haplogroup A1) with 157 identical sequences present across 16 states. Remaining haplogroups had <10 identical sequences across a maximum of three states. Some states with fewer samples had more haplogroups. Forty-one haplogroups were localized exclusively to any one state. The two most common lineages are B6 and B1 (Pangolin) whereas clade A2a (Covidex) appears to be the most predominant in India. Because the pandemic is still emerging, the observations need to be monitored.
© 2021 Laskar and Ali.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
Publication types
MeSH terms
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous