

Integrative Transkingdom Analysis of the Gut Microbiome in Antibiotic Perturbation and Critical Illness
- PMID: 33727397
- PMCID: PMC8546997
- DOI: 10.1128/mSystems.01148-20
Integrative Transkingdom Analysis of the Gut Microbiome in Antibiotic Perturbation and Critical Illness
Abstract
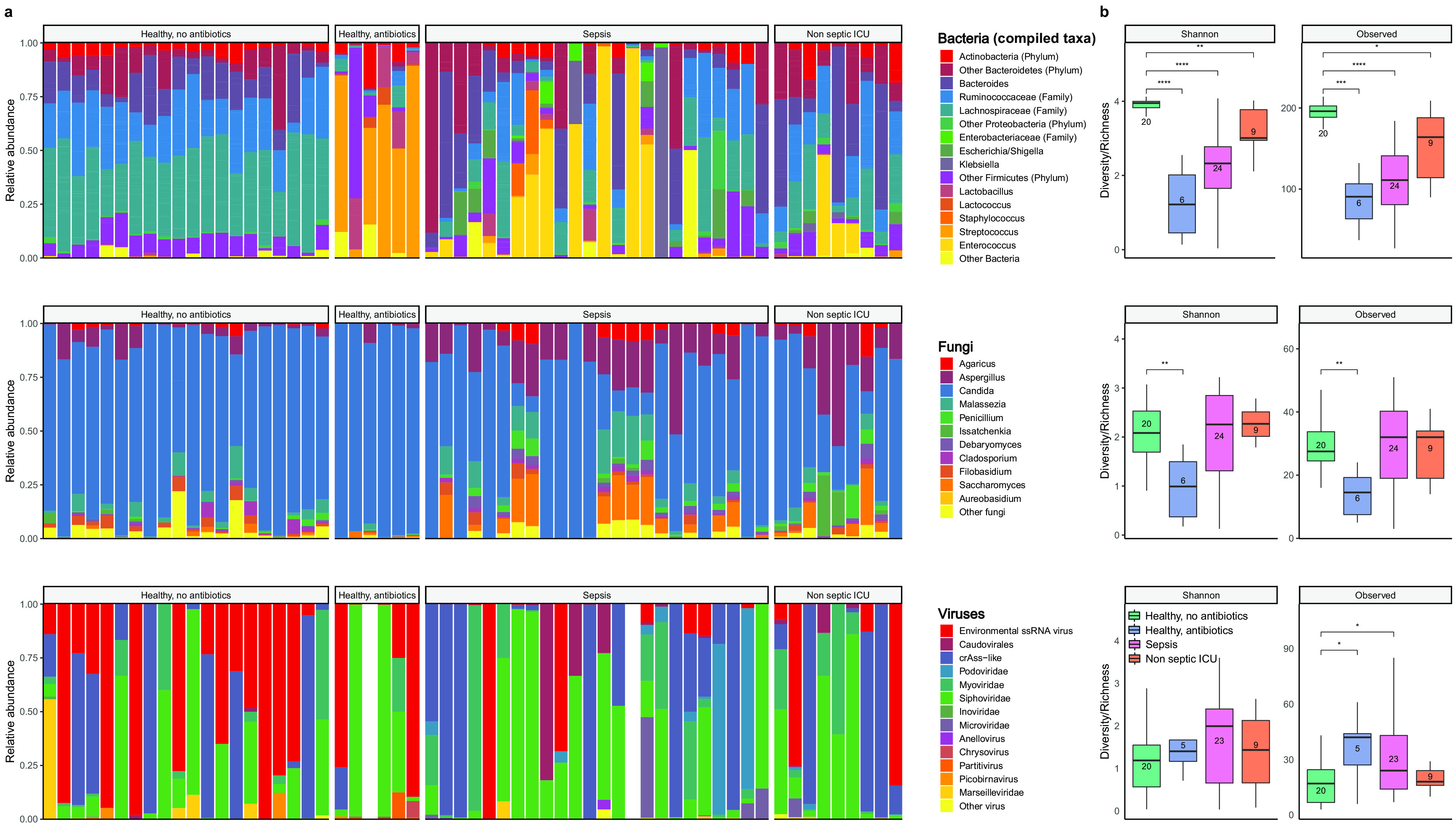
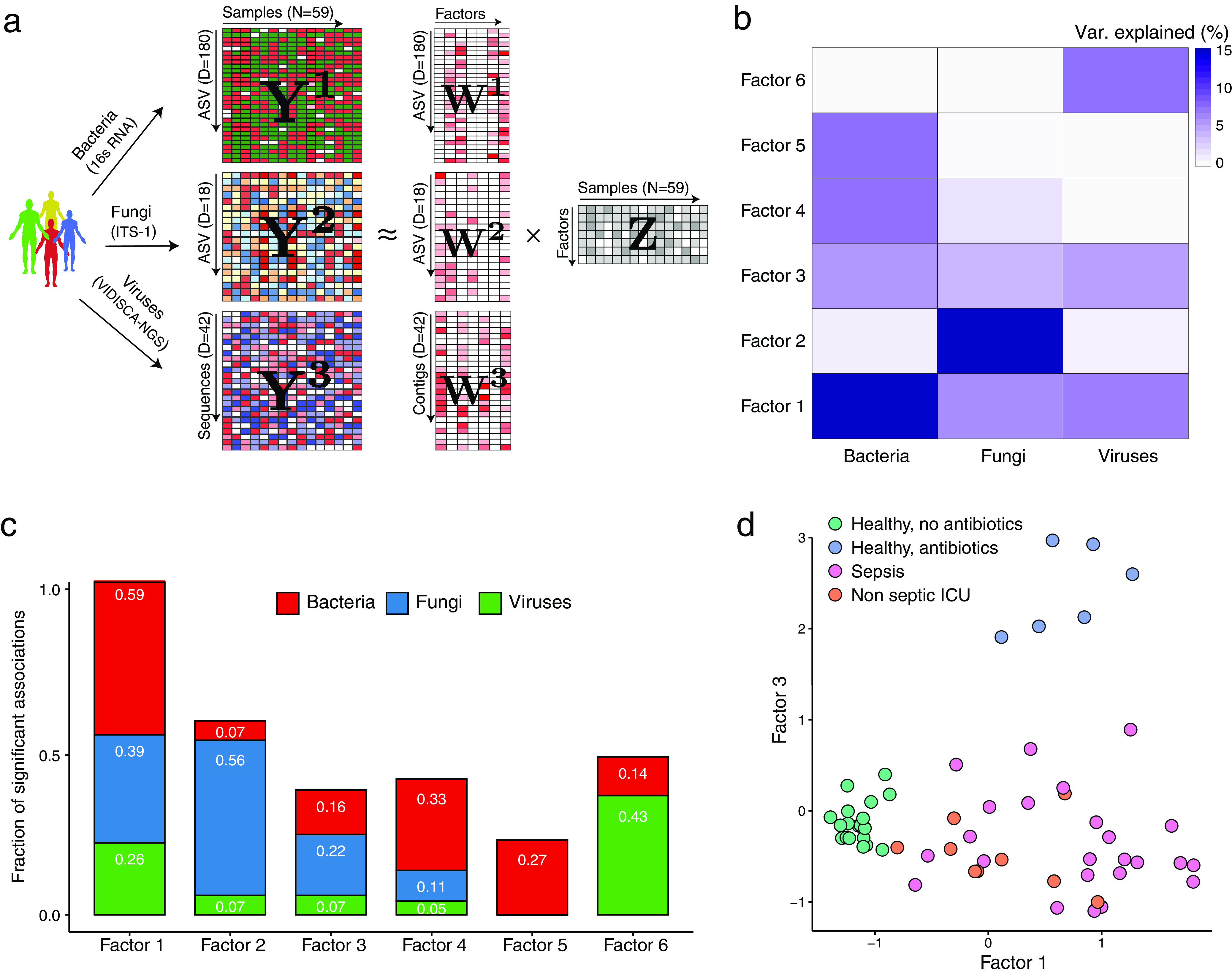
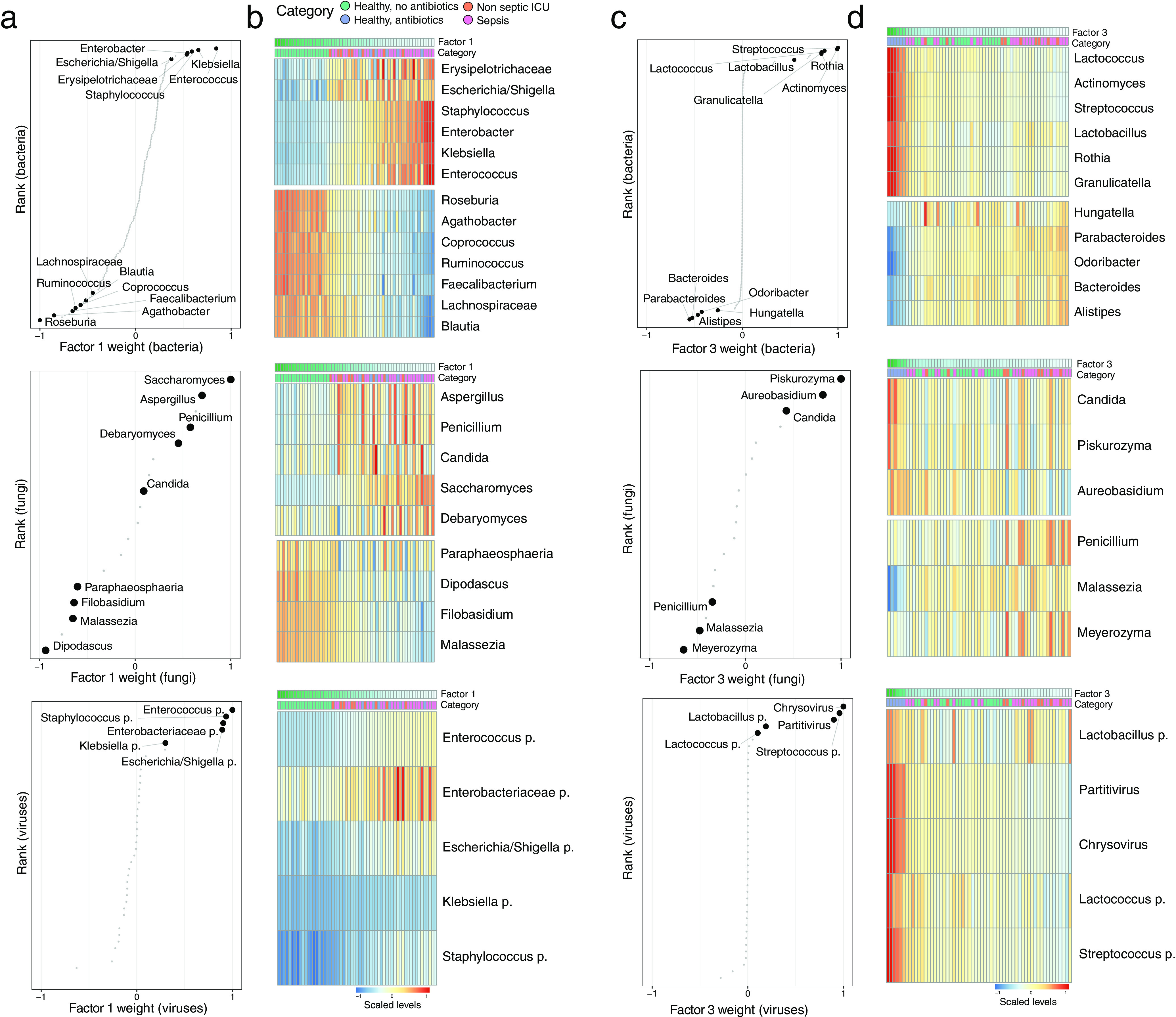
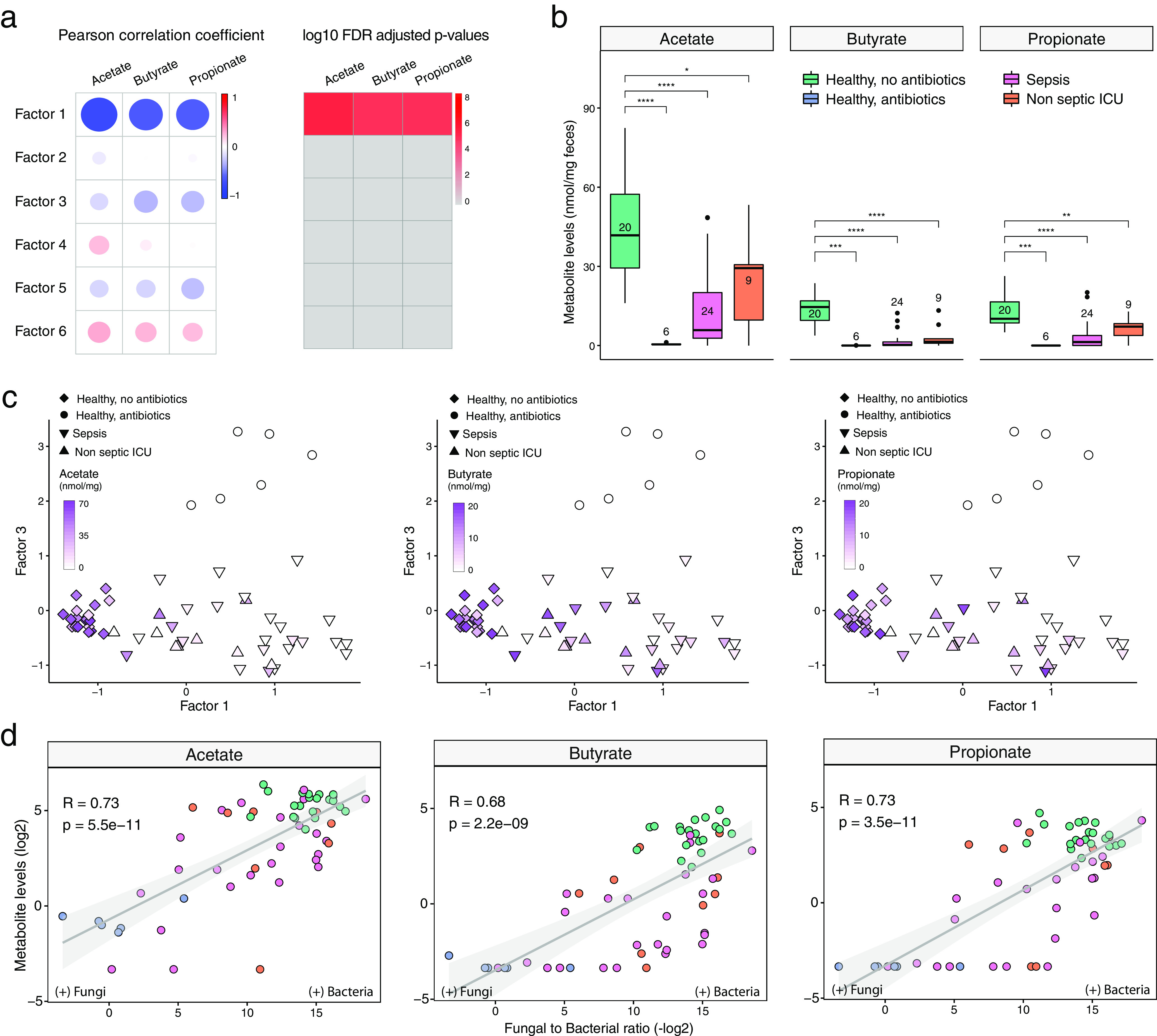
Bacterial microbiota play a critical role in mediating local and systemic immunity, and shifts in these microbial communities have been linked to impaired outcomes in critical illness. Emerging data indicate that other intestinal organisms, including bacteriophages, viruses of eukaryotes, fungi, and protozoa, are closely interlinked with the bacterial microbiota and their host, yet their collective role during antibiotic perturbation and critical illness remains to be elucidated. We employed multi-omics factor analysis (MOFA) to systematically integrate the bacterial (16S rRNA), fungal (intergenic transcribed spacer 1 rRNA), and viral (virus discovery next-generation sequencing) components of the intestinal microbiota of 33 critically ill patients with and without sepsis and 13 healthy volunteers. In addition, we quantified the absolute abundances of bacteria and fungi using 16S and 18S rRNA PCRs and characterized the short-chain fatty acids (SCFAs) butyrate, acetate, and propionate using nuclear magnetic resonance spectroscopy. We observe that a loss of the anaerobic intestinal environment is directly correlated with an overgrowth of aerobic pathobionts and their corresponding bacteriophages as well as an absolute enrichment of opportunistic yeasts capable of causing invasive disease. We also observed a strong depletion of SCFAs in both disease states, which was associated with an increased absolute abundance of fungi with respect to bacteria. Therefore, these findings illustrate the complexity of transkingdom changes following disruption of the intestinal bacterial microbiome.IMPORTANCE While numerous studies have characterized antibiotic-induced disruptions of the bacterial microbiome, few studies describe how these disruptions impact the composition of other kingdoms such as viruses, fungi, and protozoa. To address this knowledge gap, we employed MOFA to systematically integrate viral, fungal, and bacterial sequence data from critically ill patients (with and without sepsis) and healthy volunteers, both prior to and following exposure to broad-spectrum antibiotics. In doing so, we show that modulation of the bacterial component of the microbiome has implications extending beyond this kingdom alone, enabling the overgrowth of potentially invasive fungi and viruses. While numerous preclinical studies have described similar findings in vitro, we confirm these observations in humans using an integrative analytic approach. These findings underscore the potential value of multi-omics data integration tools in interrogating how different components of the microbiota contribute to disease states. In addition, our findings suggest that there is value in further studying potential adjunctive therapies using anaerobic bacteria or SCFAs to reduce fungal expansion after antibiotic exposure, which could ultimately lead to improved outcomes in the intensive care unit (ICU).
Keywords: bacteria; bacteriophages; data integration; fungi; microbiome; multi-omics.
Copyright © 2021 Haak et al.
Figures
References
-
- Schuijt TJ, Lankelma JM, Scicluna BP, de Sousa e Melo F, Roelofs JJTH, de Boer JD, Hoogendijk AJ, de Beer R, de Vos A, Belzer C, de Vos WM, van der Poll T, Wiersinga WJ. 2016. The gut microbiota plays a protective role in the host defence against pneumococcal pneumonia. Gut 65:575–583. doi:10.1136/gutjnl-2015-309728. - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
