

PPAR α Targeting GDF11 Inhibits Vascular Endothelial Cell Senescence in an Atherosclerosis Model
- PMID: 33728018
- PMCID: PMC7935606
- DOI: 10.1155/2021/2045259
PPAR α Targeting GDF11 Inhibits Vascular Endothelial Cell Senescence in an Atherosclerosis Model
Abstract
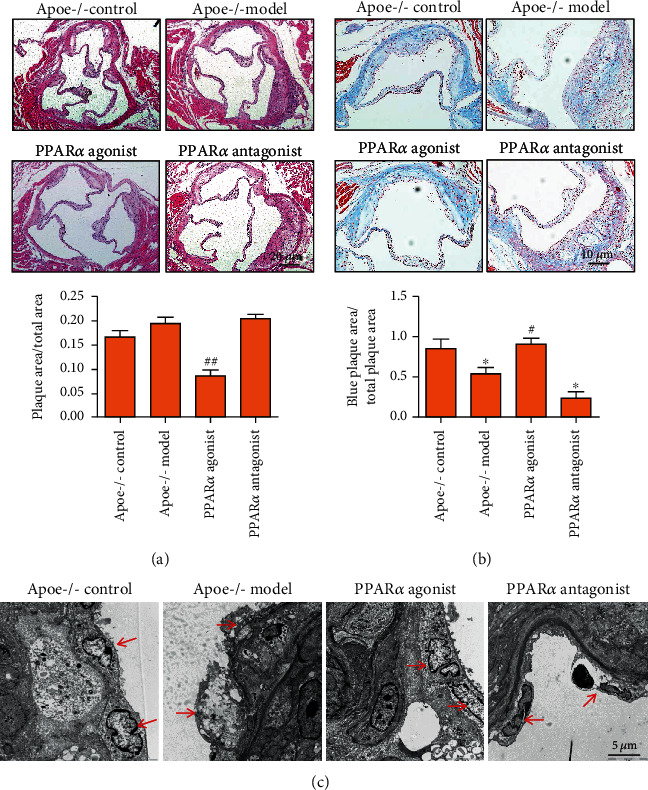
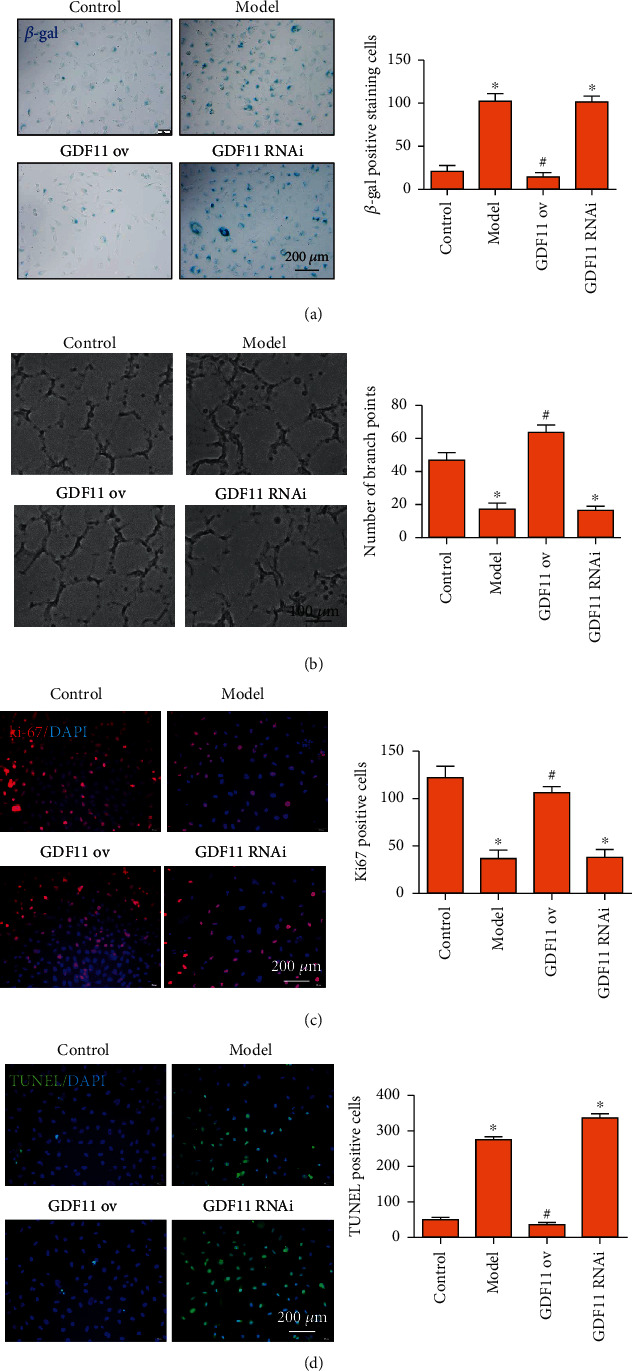
Atherosclerosis (AS) is a complex vascular disease that seriously harms the health of the elderly. It is closely related to endothelial cell aging, but the role of senescent cells in atherogenesis remains unclear. Studies have shown that peroxisome proliferator-activated receptor alpha (PPARα) inhibits the development of AS by regulating lipid metabolism. Our previous research showed that PPARα was involved in regulating the repair of damaged vascular endothelial cells. Using molecular biology and cell biology approaches to detect senescent cells in atherosclerosis-prone apolipoprotein E-deficient (Apoe -/-) mice, we found that PPARα delayed atherosclerotic plaque formation by inhibiting vascular endothelial cell senescence, which was achieved by regulating the expression of growth differentiation factor 11 (GDF11). GDF11 levels declined with age in several organs including the myocardium, bone, central nervous system, liver, and spleen in mice and participated in the regulation of aging. Our results showed that PPARα inhibited vascular endothelial cell senescence and apoptosis and promoted vascular endothelial cell proliferation and angiogenesis by increasing GDF11 production. Taken together, these results demonstrated that PPARα inhibited vascular endothelial cell aging by promoting the expression of the aging-related protein GDF11, thereby delaying the occurrence of AS.
Copyright © 2021 Fangfang Dou et al.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures




References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
