

Decrease of disease-related metabolites upon fasting in a hemizygous knock-in mouse model (Mut-ko/ki) of methylmalonic aciduria
- PMID: 33728246
- PMCID: PMC7932858
- DOI: 10.1002/jmd2.12182
Decrease of disease-related metabolites upon fasting in a hemizygous knock-in mouse model (Mut-ko/ki) of methylmalonic aciduria
Abstract
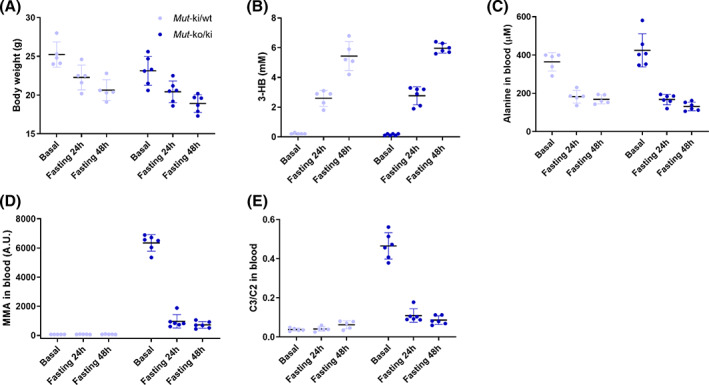
Methylmalonyl-CoA mutase (MMUT) is part of the propionyl-CoA catabolic pathway, responsible for the breakdown of branched-chain amino acids, odd-chain fatty acids and the side-chain of cholesterol. Patients with deficient activity of MMUT suffer from isolated methylmalonic aciduria (MMAuria), frequently presenting in the newborn period with failure to thrive and metabolic crisis. Even well managed patients remain at risk for metabolic crises, of which one known trigger is acute illness, which may lead to poor feeding and vomiting, putting the patient in a catabolic state. This situation is believed to result in increased breakdown of propionyl-CoA catabolic pathway precursors, producing massively elevated levels of disease related metabolites, including methylmalonic acid and propionylcarnitine. Here, we used fasting of a hemizygous mouse model (Mut-ko/ki) of MMUT deficiency to study the role of induced catabolism on metabolite production. Although mice lost weight and displayed markers consistent with a catabolic state, contrary to expectation, we found strongly reduced levels of methylmalonic acid and propionylcarnitine in fasted conditions. Switching Mut-ko/ki mice from a high-protein diet to fasted conditions, or from a standard diet to a no-protein diet, resulted in similar reductions of methylmalonic acid and propionylcarnitine levels. These results suggest, in our mouse model at least, induction of a catabolic state on its own may not be sufficient to trigger elevated metabolite levels.
Keywords: Methylmalonic aciduria; catabolism; disease amelioration; fasting; methylmalonyl‐CoA mutase; mouse model.
© 2020 The Authors. JIMD Reports published by John Wiley & Sons Ltd on behalf of SSIEM.
Conflict of interest statement
All authors declare that they have no conflicts of interest.
Figures



Similar articles
-
Novel Mouse Models of Methylmalonic Aciduria Recapitulate Phenotypic Traits with a Genetic Dosage Effect.J Biol Chem. 2016 Sep 23;291(39):20563-73. doi: 10.1074/jbc.M116.747717. Epub 2016 Aug 12. J Biol Chem. 2016. PMID: 27519416 Free PMC article.
-
In-depth phenotyping reveals common and novel disease symptoms in a hemizygous knock-in mouse model (Mut-ko/ki) of mut-type methylmalonic aciduria.Biochim Biophys Acta Mol Basis Dis. 2020 Mar 1;1866(3):165622. doi: 10.1016/j.bbadis.2019.165622. Epub 2019 Nov 23. Biochim Biophys Acta Mol Basis Dis. 2020. PMID: 31770620
-
Tricarboxylic acid cycle enzyme activities in a mouse model of methylmalonic aciduria.Mol Genet Metab. 2019 Dec;128(4):444-451. doi: 10.1016/j.ymgme.2019.10.007. Epub 2019 Oct 17. Mol Genet Metab. 2019. PMID: 31648943 Free PMC article.
-
Systematic literature review and meta-analysis on the epidemiology of methylmalonic acidemia (MMA) with a focus on MMA caused by methylmalonyl-CoA mutase (mut) deficiency.Orphanet J Rare Dis. 2019 Apr 25;14(1):84. doi: 10.1186/s13023-019-1063-z. Orphanet J Rare Dis. 2019. PMID: 31023387 Free PMC article.
-
[Methylmalonic aciduria. Classification, diagnosis and therapy (author's transl)].Klin Wochenschr. 1977 Jan 15;55(2):57-63. doi: 10.1007/BF01469083. Klin Wochenschr. 1977. PMID: 319293 Review. German.
Cited by
-
Multiomic analysis in fibroblasts of patients with inborn errors of cobalamin metabolism reveals concordance with clinical and metabolic variability.EBioMedicine. 2024 Jan;99:104911. doi: 10.1016/j.ebiom.2023.104911. Epub 2024 Jan 1. EBioMedicine. 2024. PMID: 38168585 Free PMC article.
-
Mitochondrial disease, mitophagy, and cellular distress in methylmalonic acidemia.Cell Mol Life Sci. 2021 Nov;78(21-22):6851-6867. doi: 10.1007/s00018-021-03934-3. Epub 2021 Sep 15. Cell Mol Life Sci. 2021. PMID: 34524466 Free PMC article. Review.
References
-
- Fenton WA, Gravel RA, Rosenblatt DS. Disorders of propionate and methylmalonate metabolism. In: Valle D, Beaudet A, Vogelstein B, et al., eds. The Online Metabolic and Molecular Bases of Inherited Disease. New York, NY: McGraw‐Hill Education. 2014.
-
- Rosenblatt D, Watkins D. Orphanet, Methylmalonic acidemia without homocystinuria. Orphanet, 2012. [Online]. https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=EN&Expert=293355. Accessed, June 2020.
-
- Manoli I, Venditti CP. Disorders of branched chain amino acid metabolism. Metabolic Diseases: Foundations of Clinical Management, Genetics, and Pathology. Amsterdam, The Netherlands: IOS Press; 2017:117‐135.
-
- Sbai D, Narcy C, Thompson GN, et al. Contribution of odd‐chain fatty acid oxidation to propionate production in disorders of propionate metabolism. Am J Clin Nutr. 1994;59:1332‐1337. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
