

VERSO: A comprehensive framework for the inference of robust phylogenies and the quantification of intra-host genomic diversity of viral samples
- PMID: 33728416
- PMCID: PMC7953447
- DOI: 10.1016/j.patter.2021.100212
VERSO: A comprehensive framework for the inference of robust phylogenies and the quantification of intra-host genomic diversity of viral samples
Abstract
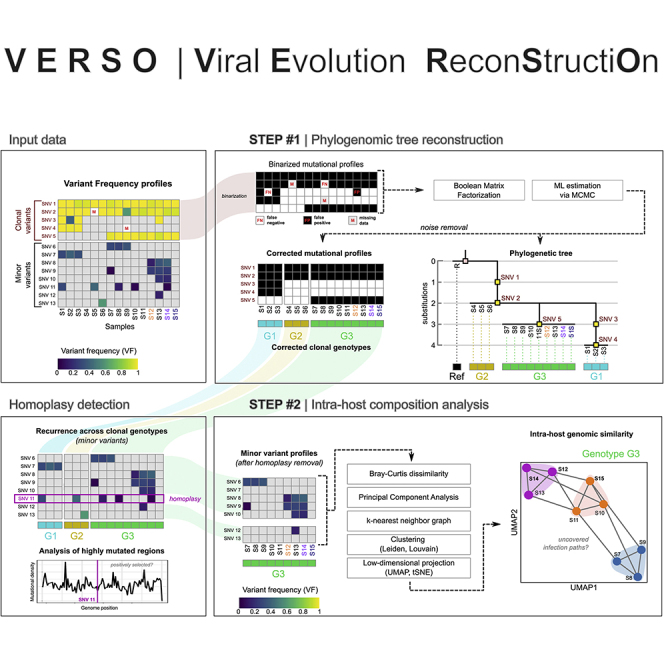
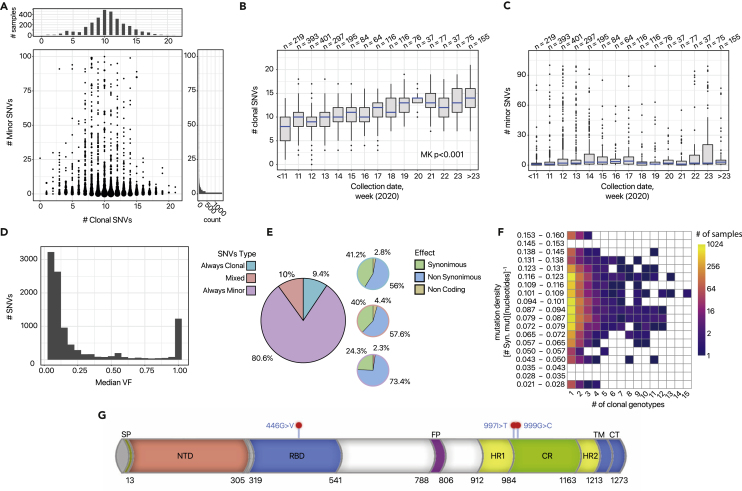
We introduce VERSO, a two-step framework for the characterization of viral evolution from sequencing data of viral genomes, which is an improvement on phylogenomic approaches for consensus sequences. VERSO exploits an efficient algorithmic strategy to return robust phylogenies from clonal variant profiles, also in conditions of sampling limitations. It then leverages variant frequency patterns to characterize the intra-host genomic diversity of samples, revealing undetected infection chains and pinpointing variants likely involved in homoplasies. On simulations, VERSO outperforms state-of-the-art tools for phylogenetic inference. Notably, the application to 6,726 amplicon and RNA sequencing samples refines the estimation of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) evolution, while co-occurrence patterns of minor variants unveil undetected infection paths, which are validated with contact tracing data. Finally, the analysis of SARS-CoV-2 mutational landscape uncovers a temporal increase of overall genomic diversity and highlights variants transiting from minor to clonal state and homoplastic variants, some of which fall on the spike gene. Available at: https://github.com/BIMIB-DISCo/VERSO.
Keywords: COVID-19; SARS-CoV-2; genomic surveillance; intra-host genomic diversity; phylogenomics; viral evolution; viral variants.
© 2021 The Authors.
Conflict of interest statement
The authors declare no competing interests.
Figures




Similar articles
-
Mutational signatures and heterogeneous host response revealed via large-scale characterization of SARS-CoV-2 genomic diversity.iScience. 2021 Feb 19;24(2):102116. doi: 10.1016/j.isci.2021.102116. Epub 2021 Jan 28. iScience. 2021. PMID: 33532709 Free PMC article.
-
SARS Coronavirus-2 variant tracing within the first Coronavirus Disease 19 clusters in northern Germany.Clin Microbiol Infect. 2021 Jan;27(1):130.e5-130.e8. doi: 10.1016/j.cmi.2020.09.034. Epub 2020 Sep 29. Clin Microbiol Infect. 2021. PMID: 33007476 Free PMC article.
-
Optimized quantification of intra-host viral diversity in SARS-CoV-2 and influenza virus sequence data.mBio. 2023 Aug 31;14(4):e0104623. doi: 10.1128/mbio.01046-23. Epub 2023 Jun 30. mBio. 2023. PMID: 37389439 Free PMC article.
-
Improved SARS-CoV-2 sequencing surveillance allows the identification of new variants and signatures in infected patients.Genome Med. 2022 Aug 12;14(1):90. doi: 10.1186/s13073-022-01098-8. Genome Med. 2022. PMID: 35962405 Free PMC article.
-
Intra-host evolution during SARS-CoV-2 prolonged infection.Virus Evol. 2021 Sep 29;7(2):veab078. doi: 10.1093/ve/veab078. Virus Evol. 2021. PMID: 34642605 Free PMC article.
Cited by
-
Early detection and improved genomic surveillance of SARS-CoV-2 variants from deep sequencing data.iScience. 2022 Jun 17;25(6):104487. doi: 10.1016/j.isci.2022.104487. Epub 2022 May 30. iScience. 2022. PMID: 35677393 Free PMC article.
-
Unveiling Inter- and Intra-Patient Sequence Variability with a Multi-Sample Coronavirus Target Enrichment Approach.Viruses. 2024 May 15;16(5):786. doi: 10.3390/v16050786. Viruses. 2024. PMID: 38793667 Free PMC article.
-
Transmission of SARS-CoV-2 in domestic cats imposes a narrow bottleneck.PLoS Pathog. 2021 Feb 26;17(2):e1009373. doi: 10.1371/journal.ppat.1009373. eCollection 2021 Feb. PLoS Pathog. 2021. PMID: 33635912 Free PMC article.
-
Transmission of SARS-CoV-2 in domestic cats imposes a narrow bottleneck.bioRxiv [Preprint]. 2021 Jan 4:2020.11.16.384917. doi: 10.1101/2020.11.16.384917. bioRxiv. 2021. Update in: PLoS Pathog. 2021 Feb 26;17(2):e1009373. doi: 10.1371/journal.ppat.1009373. PMID: 33236011 Free PMC article. Updated. Preprint.
-
Non-uniform aspects of the SARS-CoV-2 intraspecies evolution reopen question of its origin.Int J Biol Macromol. 2022 Dec 1;222(Pt A):972-993. doi: 10.1016/j.ijbiomac.2022.09.184. Epub 2022 Sep 26. Int J Biol Macromol. 2022. PMID: 36174872 Free PMC article.
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
