

Coordinated signaling of activating transcription factor 6α and inositol-requiring enzyme 1α regulates hepatic stellate cell-mediated fibrogenesis in mice
- PMID: 33728997
- PMCID: PMC8202196
- DOI: 10.1152/ajpgi.00453.2020
Coordinated signaling of activating transcription factor 6α and inositol-requiring enzyme 1α regulates hepatic stellate cell-mediated fibrogenesis in mice
Abstract
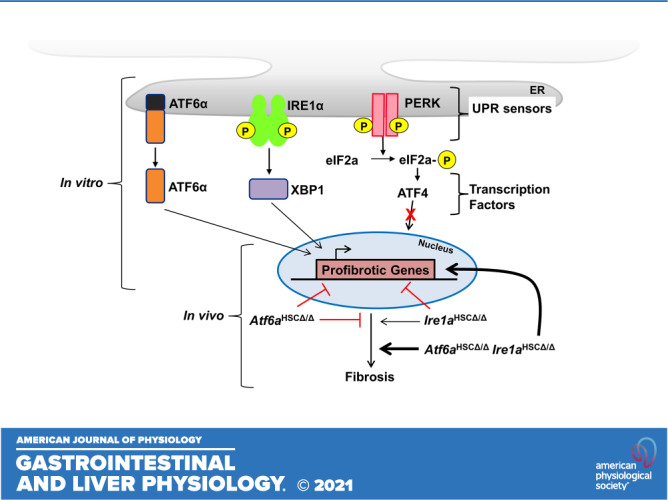
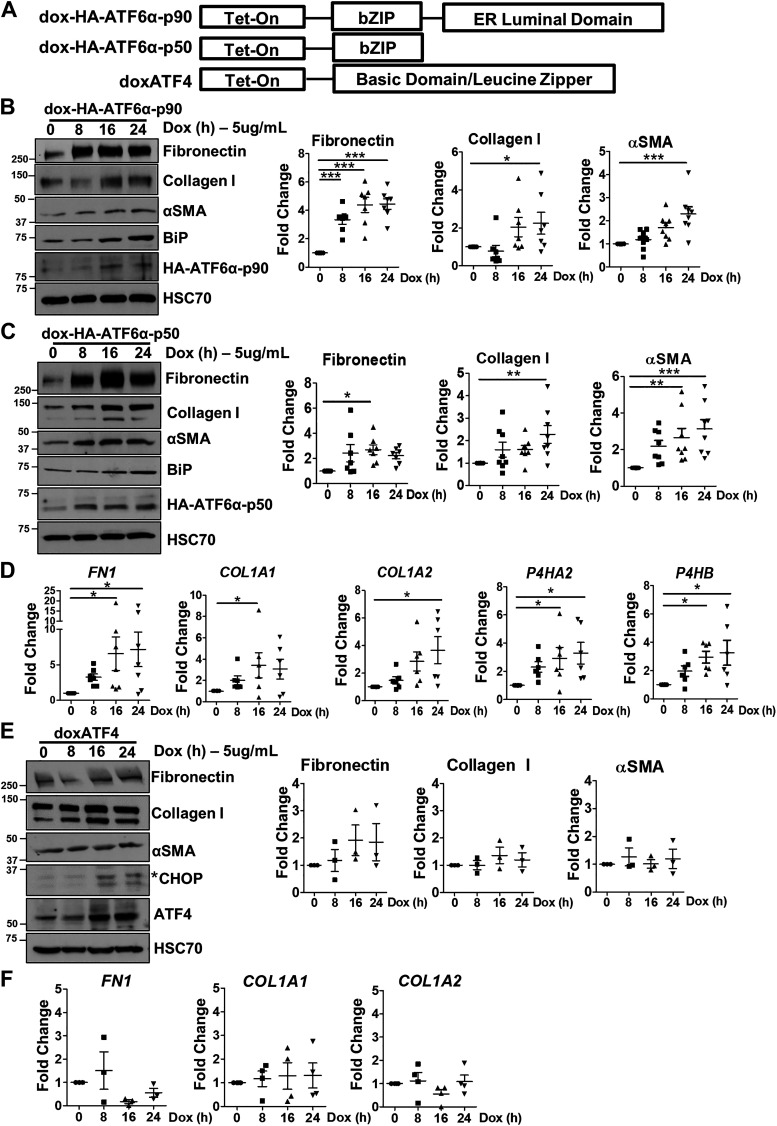
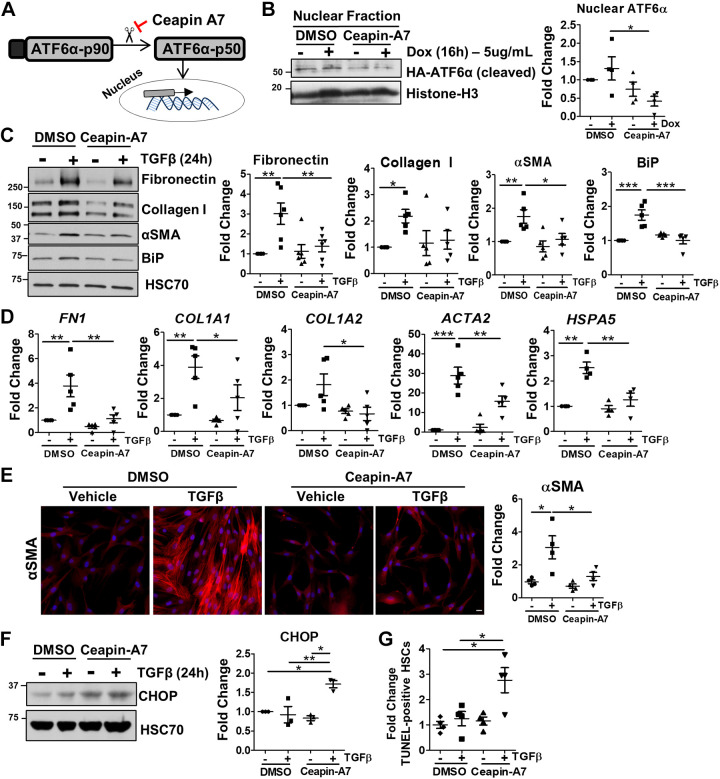
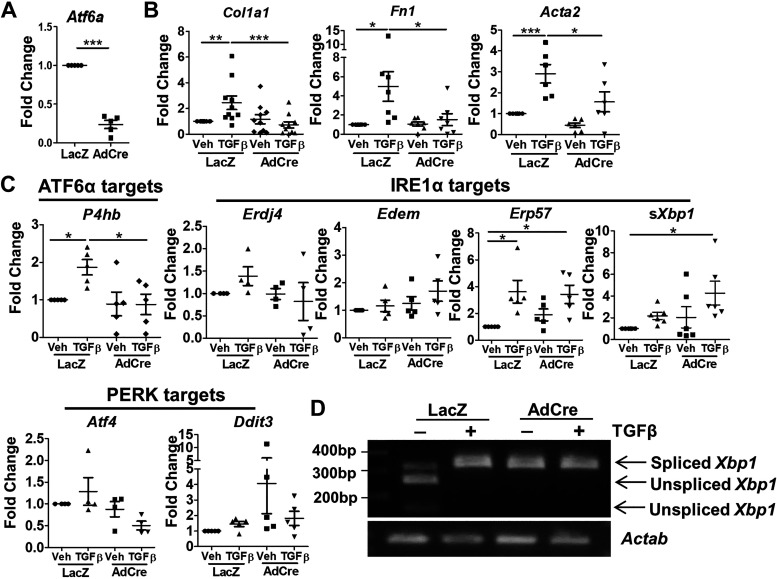
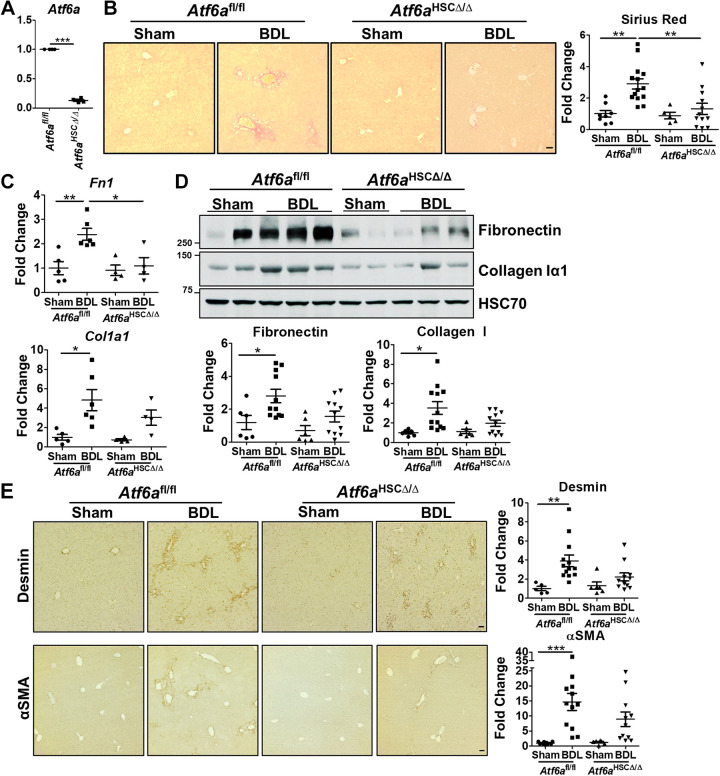
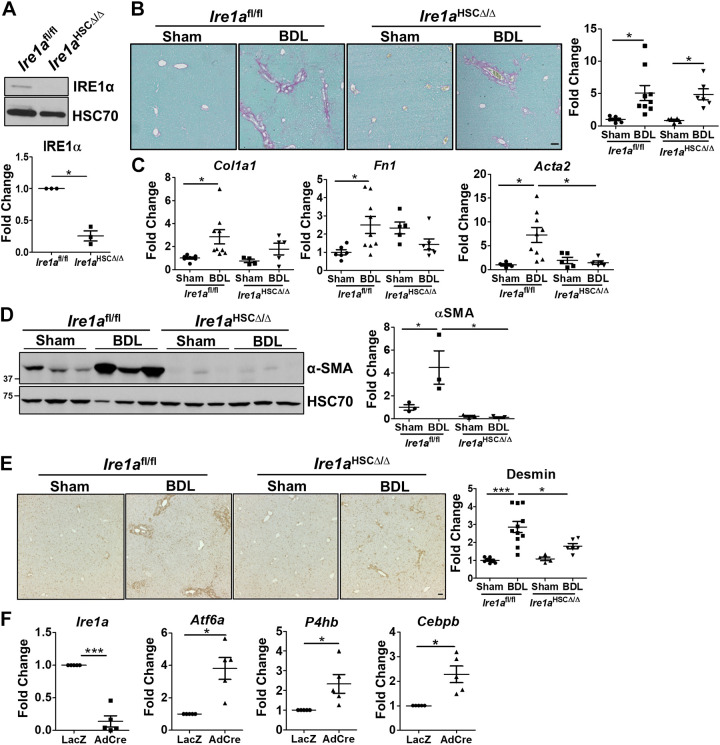
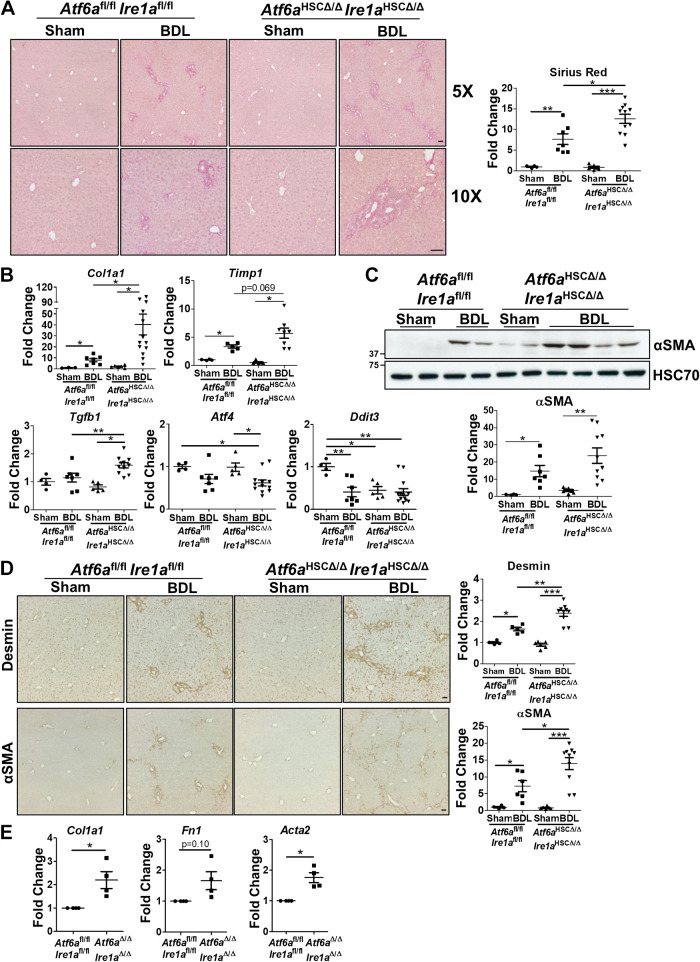
Liver injury and the unfolded protein response (UPR) are tightly linked, but their relationship differs with cell type and injurious stimuli. UPR initiation promotes hepatic stellate cell (HSC) activation and fibrogenesis, but the underlying mechanisms are unclear. Despite the complexity and overlap downstream of UPR transducers inositol-requiring protein 1α (IRE1α), activating transcription factor 6α (ATF6α), and protein kinase RNA-like ER kinase (PERK), previous research in HSCs primarily focused on IRE1α. Here, we investigated the fibrogenic role of ATF6α or PERK in vitro and HSC-specific UPR signaling in vivo. Overexpression of ATF6α, but not the PERK effector activating transcription factor 4 (ATF4), promoted HSC activation and fibrogenic gene transcription in immortalized HSCs. Furthermore, ATF6α inhibition through Ceapin-A7, or Atf6a deletion, disrupted transforming growth factor β (TGFβ)-mediated activation of primary human hepatic stellate cells (hHSCs) or murine hepatic stellate cells (mHSCs), respectively. We investigated the fibrogenic role of ATF6α in vivo through conditional HSC-specific Atf6a deletion. Atf6aHSCΔ/Δ mice displayed reduced fibrosis and HSC activation following bile duct ligation (BDL) or carbon tetrachloride (CCl4)-induced injury. The Atf6aHSCΔ/Δ phenotype differed from HSC-specific Ire1a deletion, as Ire1aHSCΔ/Δ mice showed reduced fibrogenic gene transcription but no changes in fibrosis compared with Ire1afl/fl mice following BDL. Interestingly, ATF6α signaling increased in Ire1aΔ/Δ HSCs, whereas IRE1α signaling was upregulated in Atf6aΔ/Δ HSCs. Finally, we asked whether co-deletion of Atf6a and Ire1a additively limits fibrosis. Unexpectedly, fibrosis worsened in Atf6aHSCΔ/ΔIre1aHSCΔ/Δ mice following BDL, and Atf6aΔ/ΔIre1aΔ/Δ mHSCs showed increased fibrogenic gene transcription. ATF6α and IRE1α individually promote fibrogenic transcription in HSCs, and ATF6α drives fibrogenesis in vivo. Unexpectedly, disruption of both pathways sensitizes the liver to fibrogenesis, suggesting that fine-tuned UPR signaling is critical for regulating HSC activation and fibrogenesis.NEW & NOTEWORTHY ATF6α is a critical driver of hepatic stellate cell (HSC) activation in vitro. HSC-specific deletion of Atf6a limits fibrogenesis in vivo despite increased IRE1α signaling. Conditional deletion of Ire1α from HSCs limits fibrogenic gene transcription without impacting overall fibrosis. This could be due in part to observed upregulation of the ATF6α pathway. Dual loss of Atf6a and Ire1a from HSCs worsens fibrosis in vivo through enhanced HSC activation.
Keywords: ER stress; endoplasmic reticulum; fibrosis; hepatic fibrosis; unfolded protein response.
Conflict of interest statement
No conflicts of interest, financial or otherwise, are declared by the authors.
Figures
Similar articles
-
Stress and Liver Fibrogenesis: Understanding the Role and Regulation of Stress Response Pathways in Hepatic Stellate Cells.Am J Pathol. 2023 Oct;193(10):1363-1376. doi: 10.1016/j.ajpath.2023.06.006. Epub 2023 Jul 7. Am J Pathol. 2023. PMID: 37422148 Free PMC article. Review.
-
Transforming growth factor β (TGFβ) cross-talk with the unfolded protein response is critical for hepatic stellate cell activation.J Biol Chem. 2019 Mar 1;294(9):3137-3151. doi: 10.1074/jbc.RA118.005761. Epub 2019 Jan 4. J Biol Chem. 2019. PMID: 30610118 Free PMC article.
-
Gli2-regulated activation of hepatic stellate cells and liver fibrosis by TGF-β signaling.Am J Physiol Gastrointest Liver Physiol. 2021 May 1;320(5):G720-G728. doi: 10.1152/ajpgi.00310.2020. Epub 2021 Mar 17. Am J Physiol Gastrointest Liver Physiol. 2021. PMID: 33728992
-
Hepatic stellate cell autophagy inhibits extracellular vesicle release to attenuate liver fibrosis.J Hepatol. 2020 Nov;73(5):1144-1154. doi: 10.1016/j.jhep.2020.04.044. Epub 2020 May 8. J Hepatol. 2020. PMID: 32389810 Free PMC article.
-
Liver Fibrosis: From Basic Science towards Clinical Progress, Focusing on the Central Role of Hepatic Stellate Cells.Int J Mol Sci. 2024 Jul 18;25(14):7873. doi: 10.3390/ijms25147873. Int J Mol Sci. 2024. PMID: 39063116 Free PMC article. Review.
Cited by
-
QRICH1 regulates ATF6 transcription to affect pathological cardiac hypertrophy progression.Mol Med. 2025 May 13;31(1):183. doi: 10.1186/s10020-025-01241-2. Mol Med. 2025. PMID: 40355839 Free PMC article.
-
Endoplasmic reticulum stress sensor protein PERK in hepatic stellate cells promotes the progression of hepatocellular carcinoma via p38δ MAPK/IL-1β axis.Sci Rep. 2025 Jun 6;15(1):20030. doi: 10.1038/s41598-025-04150-w. Sci Rep. 2025. PMID: 40481076 Free PMC article.
-
ER Disposal Pathways in Chronic Liver Disease: Protective, Pathogenic, and Potential Therapeutic Targets.Front Mol Biosci. 2022 Jan 31;8:804097. doi: 10.3389/fmolb.2021.804097. eCollection 2021. Front Mol Biosci. 2022. PMID: 35174209 Free PMC article. Review.
-
Natural compound Alternol actives multiple endoplasmic reticulum stress-responding pathways contributing to cell death.Front Pharmacol. 2024 May 20;15:1397116. doi: 10.3389/fphar.2024.1397116. eCollection 2024. Front Pharmacol. 2024. PMID: 38831880 Free PMC article.
-
Stress and Liver Fibrogenesis: Understanding the Role and Regulation of Stress Response Pathways in Hepatic Stellate Cells.Am J Pathol. 2023 Oct;193(10):1363-1376. doi: 10.1016/j.ajpath.2023.06.006. Epub 2023 Jul 7. Am J Pathol. 2023. PMID: 37422148 Free PMC article. Review.
References
-
- Iwaisako K, Jiang C, Zhang M, Cong M, Moore-Morris TJ, Park TJ, Liu X, Xu J, Wang P, Paik YH, Meng F, Asagiri M, Murray LA, Hofmann AF, Iida T, Glass CK, Brenner DA, Kisseleva T. Origin of myofibroblasts in the fibrotic liver in mice. Proc Natl Acad Sci USA 111: E3297–E3305, 2014. doi:10.1073/pnas.1400062111. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials