

TorchMD: A Deep Learning Framework for Molecular Simulations
- PMID: 33729795
- PMCID: PMC8486166
- DOI: 10.1021/acs.jctc.0c01343
TorchMD: A Deep Learning Framework for Molecular Simulations
Abstract
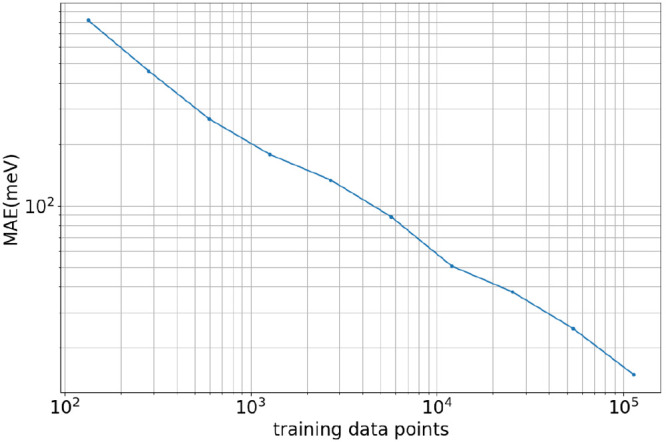
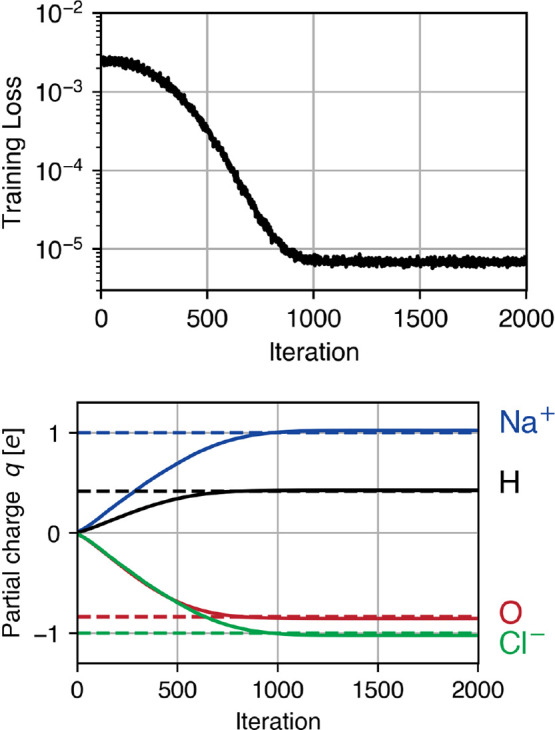
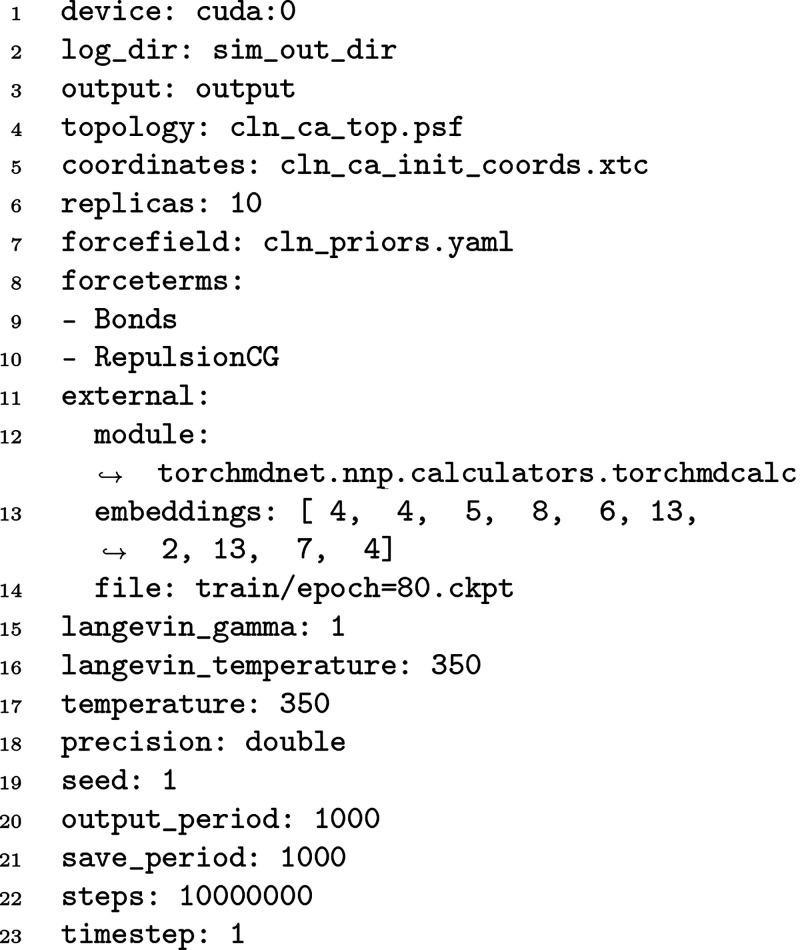
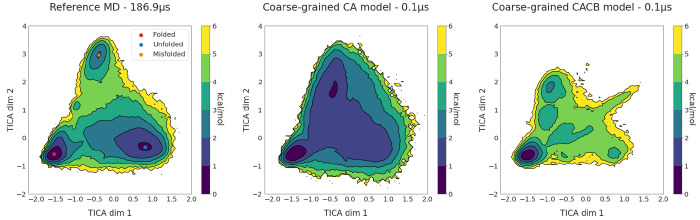
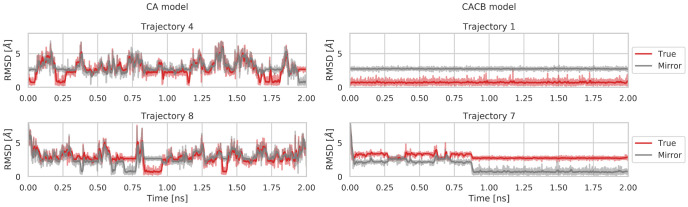
Molecular dynamics simulations provide a mechanistic description of molecules by relying on empirical potentials. The quality and transferability of such potentials can be improved leveraging data-driven models derived with machine learning approaches. Here, we present TorchMD, a framework for molecular simulations with mixed classical and machine learning potentials. All force computations including bond, angle, dihedral, Lennard-Jones, and Coulomb interactions are expressed as PyTorch arrays and operations. Moreover, TorchMD enables learning and simulating neural network potentials. We validate it using standard Amber all-atom simulations, learning an ab initio potential, performing an end-to-end training, and finally learning and simulating a coarse-grained model for protein folding. We believe that TorchMD provides a useful tool set to support molecular simulations of machine learning potentials. Code and data are freely available at github.com/torchmd.
Conflict of interest statement
The authors declare no competing financial interest.
Figures


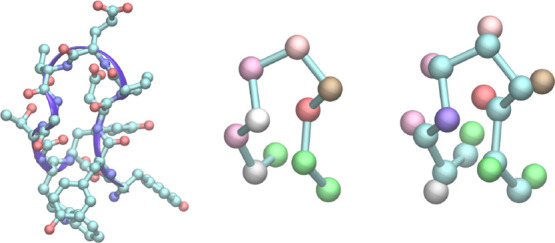
References
-
- Schütt K.; Kindermans P.-J.; Felix H. E. S.; Chmiela S.; Tkatchenko A.; Müller K.-R. Schnet: A continuous-filter convolutional neural network for modeling quantum interactions. Advances in neural information processing systems 2017, 991–1001.
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases