

Defining the underlying defect in insulin action in type 2 diabetes
- PMID: 33730188
- PMCID: PMC8916220
- DOI: 10.1007/s00125-021-05415-5
Defining the underlying defect in insulin action in type 2 diabetes
Erratum in
-
Correction to: Defining the underlying defect in insulin action in type 2 diabetes.Diabetologia. 2022 Jun;65(6):1064. doi: 10.1007/s00125-022-05684-8. Diabetologia. 2022. PMID: 35320373 No abstract available.
Abstract
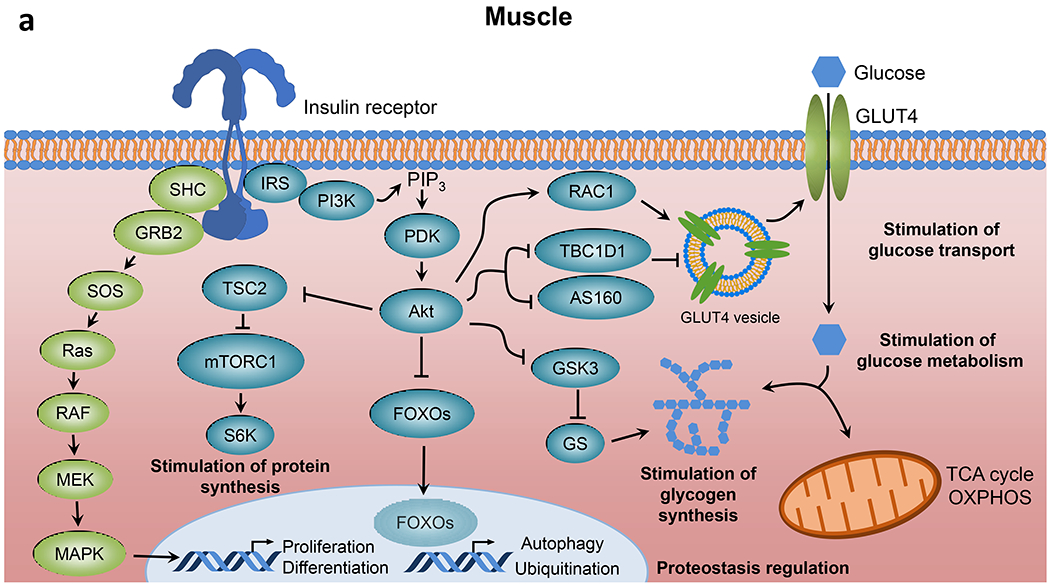
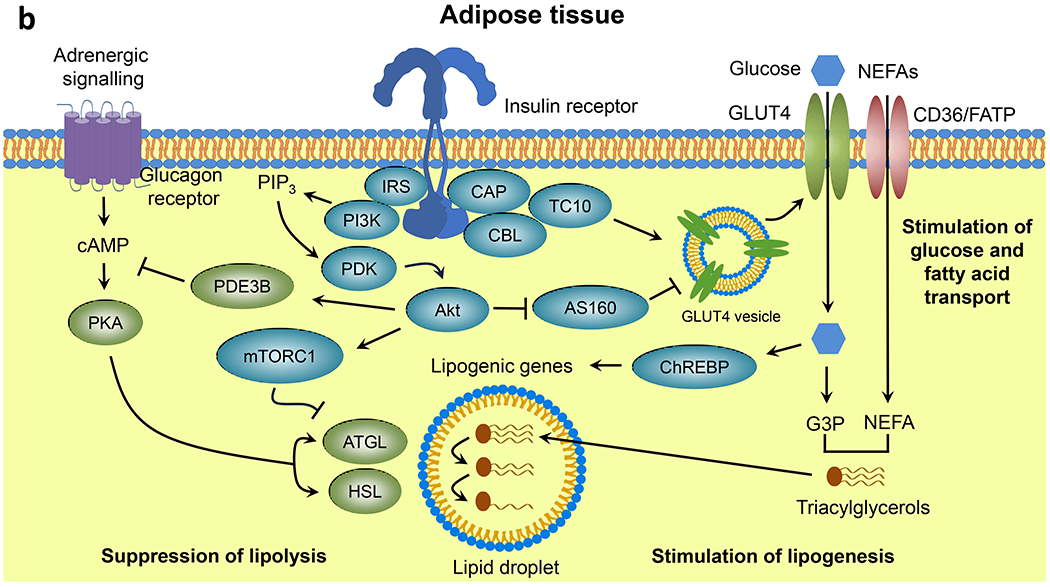
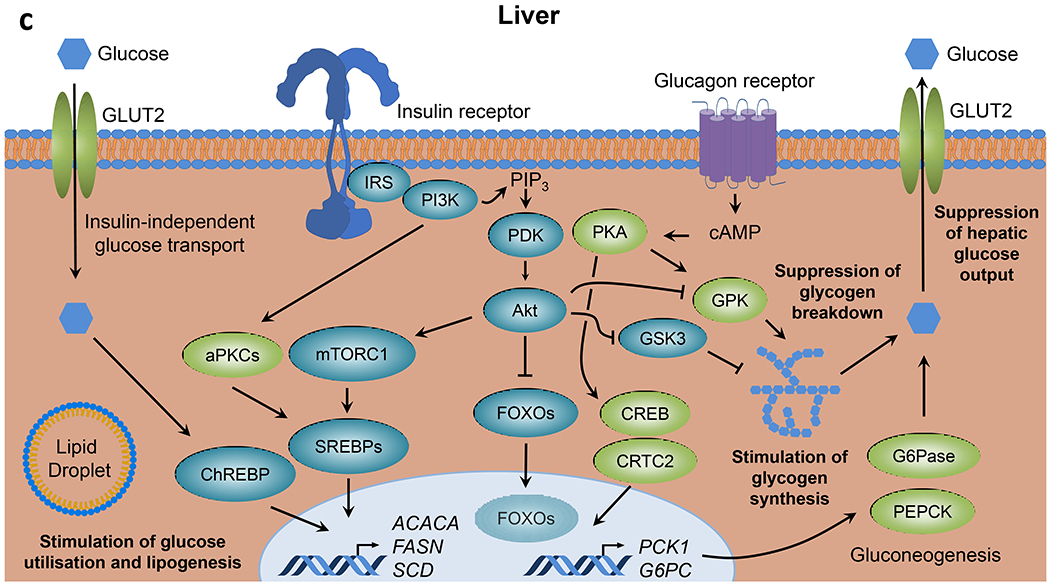
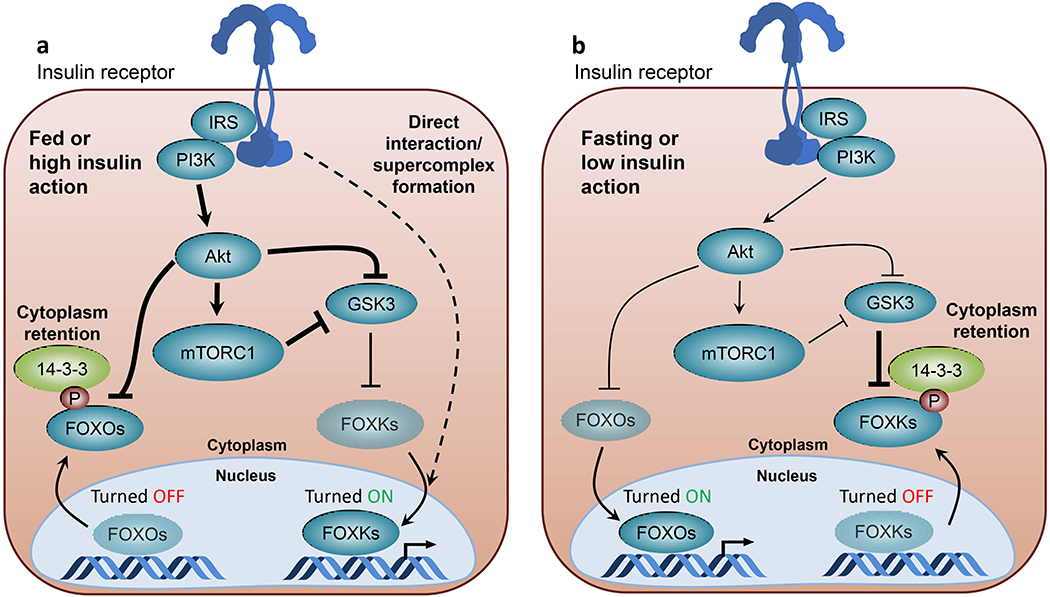
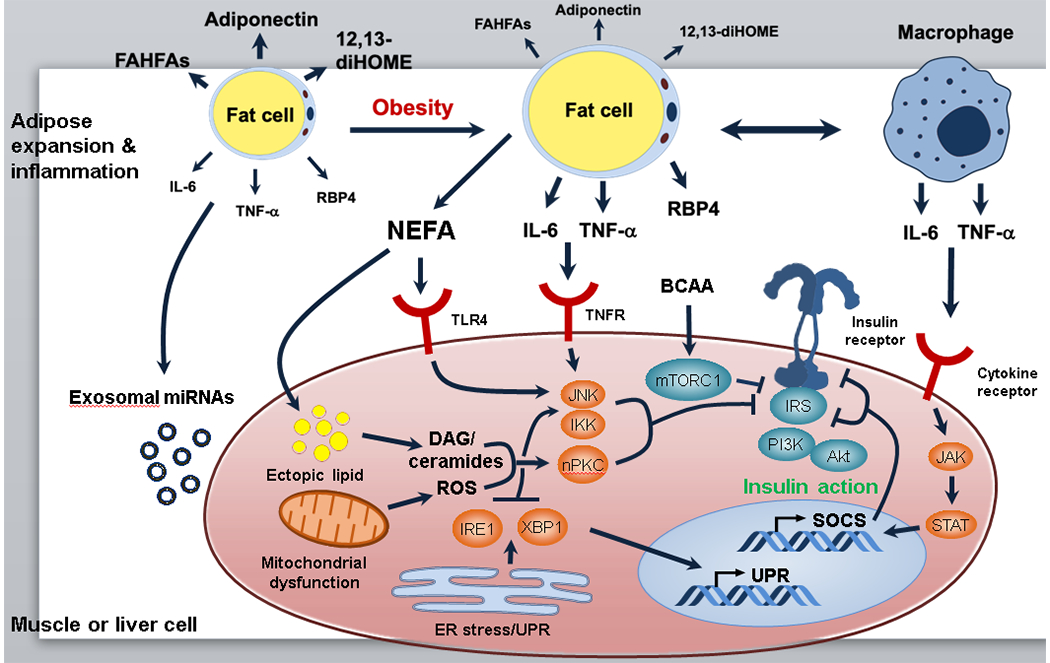
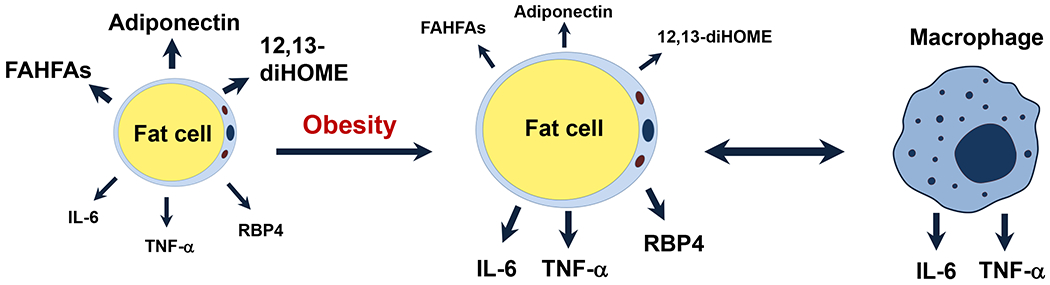
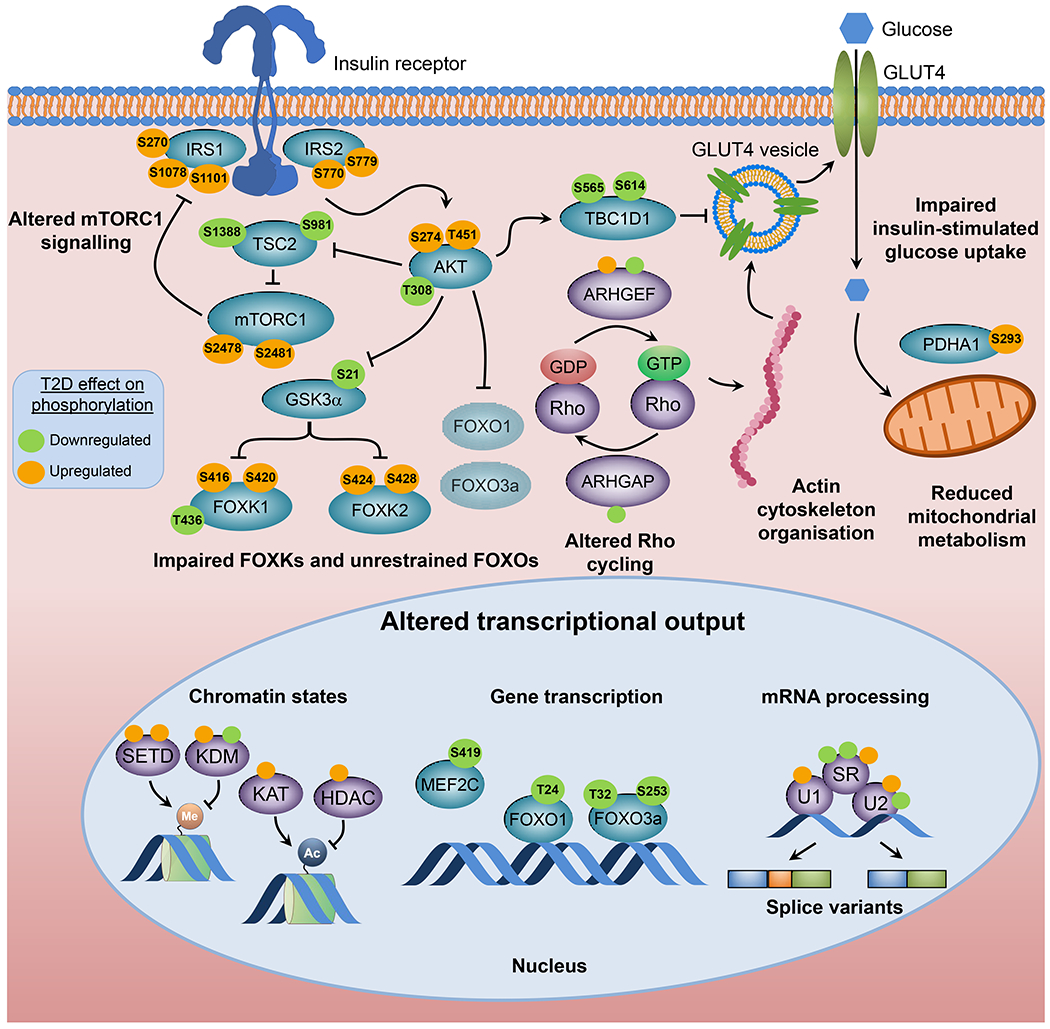
Insulin resistance is one of the earliest defects in the pathogenesis of type 2 diabetes. Over the past 50 years, elucidation of the insulin signalling network has provided important mechanistic insights into the abnormalities of glucose, lipid and protein metabolism that underlie insulin resistance. In classical target tissues (liver, muscle and adipose tissue), insulin binding to its receptor initiates a broad signalling cascade mediated by changes in phosphorylation, gene expression and vesicular trafficking that result in increased nutrient utilisation and storage, and suppression of catabolic processes. Insulin receptors are also expressed in non-classical targets, such as the brain and endothelial cells, where it helps regulate appetite, energy expenditure, reproductive hormones, mood/behaviour and vascular function. Recent progress in cell biology and unbiased molecular profiling by mass spectrometry and DNA/RNA-sequencing has provided a unique opportunity to dissect the determinants of insulin resistance in type 2 diabetes and the metabolic syndrome; best studied are extrinsic factors, such as circulating lipids, amino acids and other metabolites and exosomal microRNAs. More challenging has been defining the cell-intrinsic factors programmed by genetics and epigenetics that underlie insulin resistance. In this regard, studies using human induced pluripotent stem cells and tissues point to cell-autonomous alterations in signalling super-networks, involving changes in phosphorylation and gene expression both inside and outside the canonical insulin signalling pathway. Understanding how these multi-layered molecular networks modulate insulin action and metabolism in different tissues will open new avenues for therapy and prevention of type 2 diabetes and its associated pathologies.
Keywords: Cell-autonomous; Insulin action; Insulin resistance; Phosphorylation; Review; The metabolic syndrome; Tissue crosstalk; Type 2 diabetes; iPS cells.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
