

H3K27ac bookmarking promotes rapid post-mitotic activation of the pluripotent stem cell program without impacting 3D chromatin reorganization
- PMID: 33730542
- PMCID: PMC8052294
- DOI: 10.1016/j.molcel.2021.02.032
H3K27ac bookmarking promotes rapid post-mitotic activation of the pluripotent stem cell program without impacting 3D chromatin reorganization
Abstract
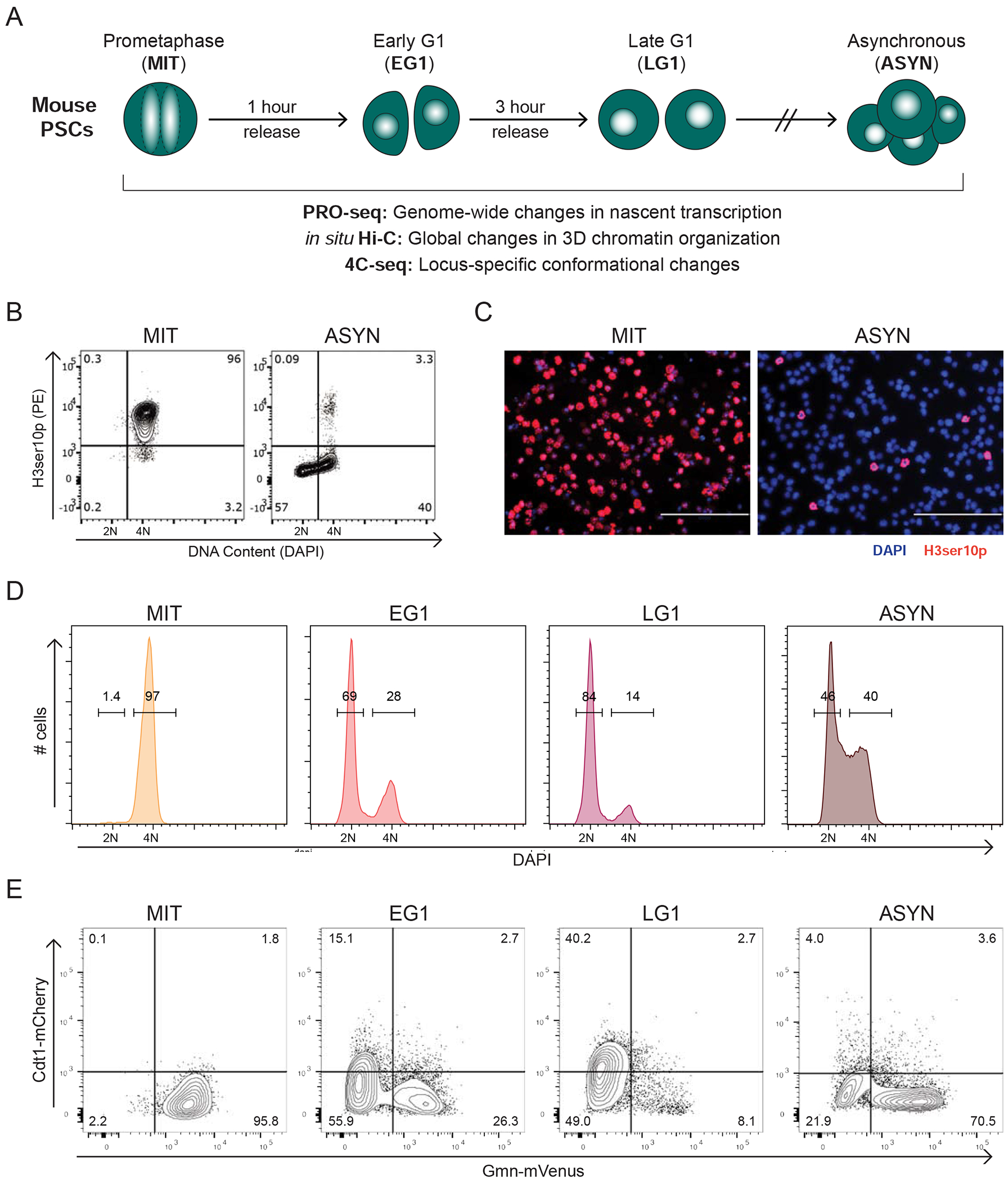
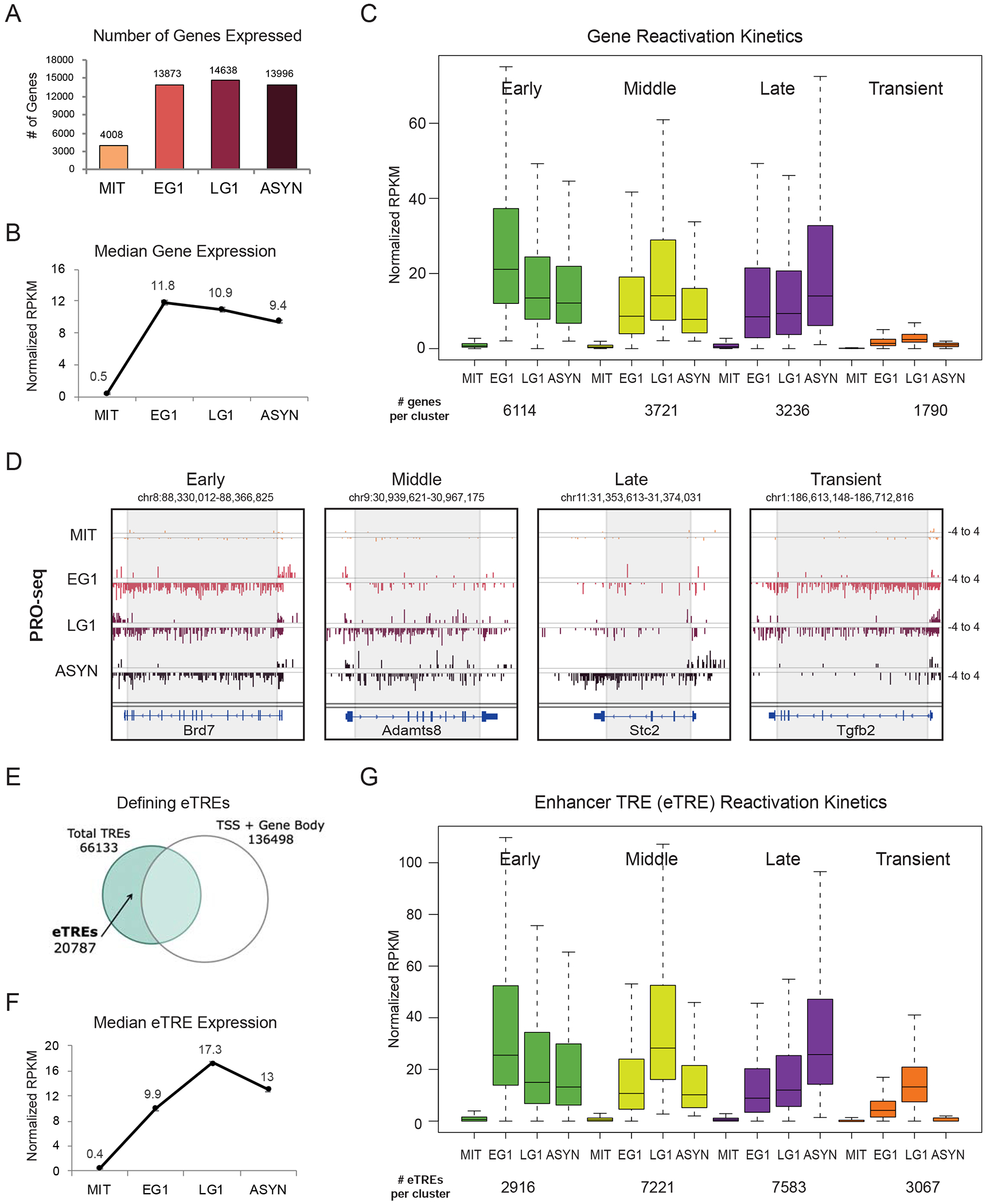
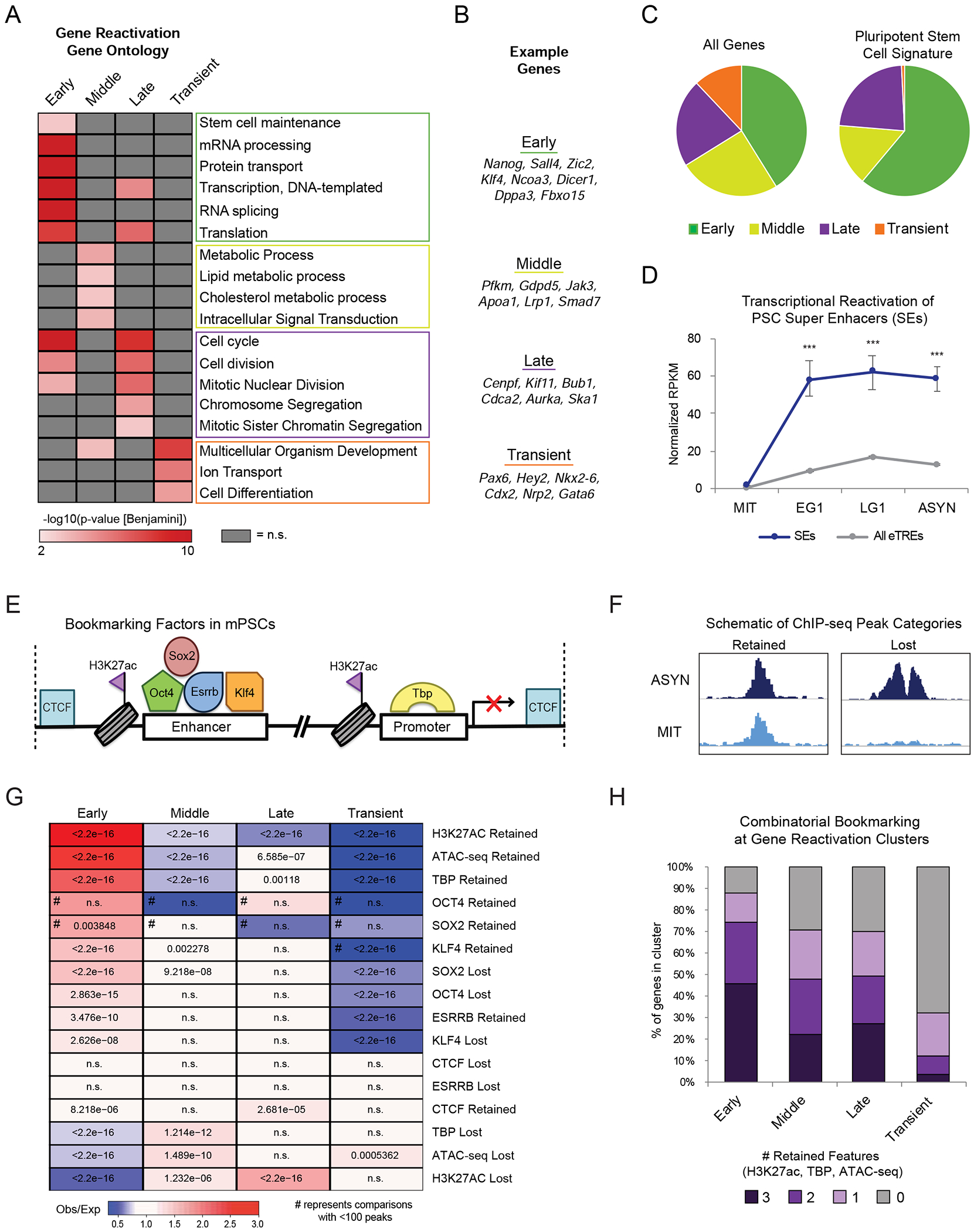
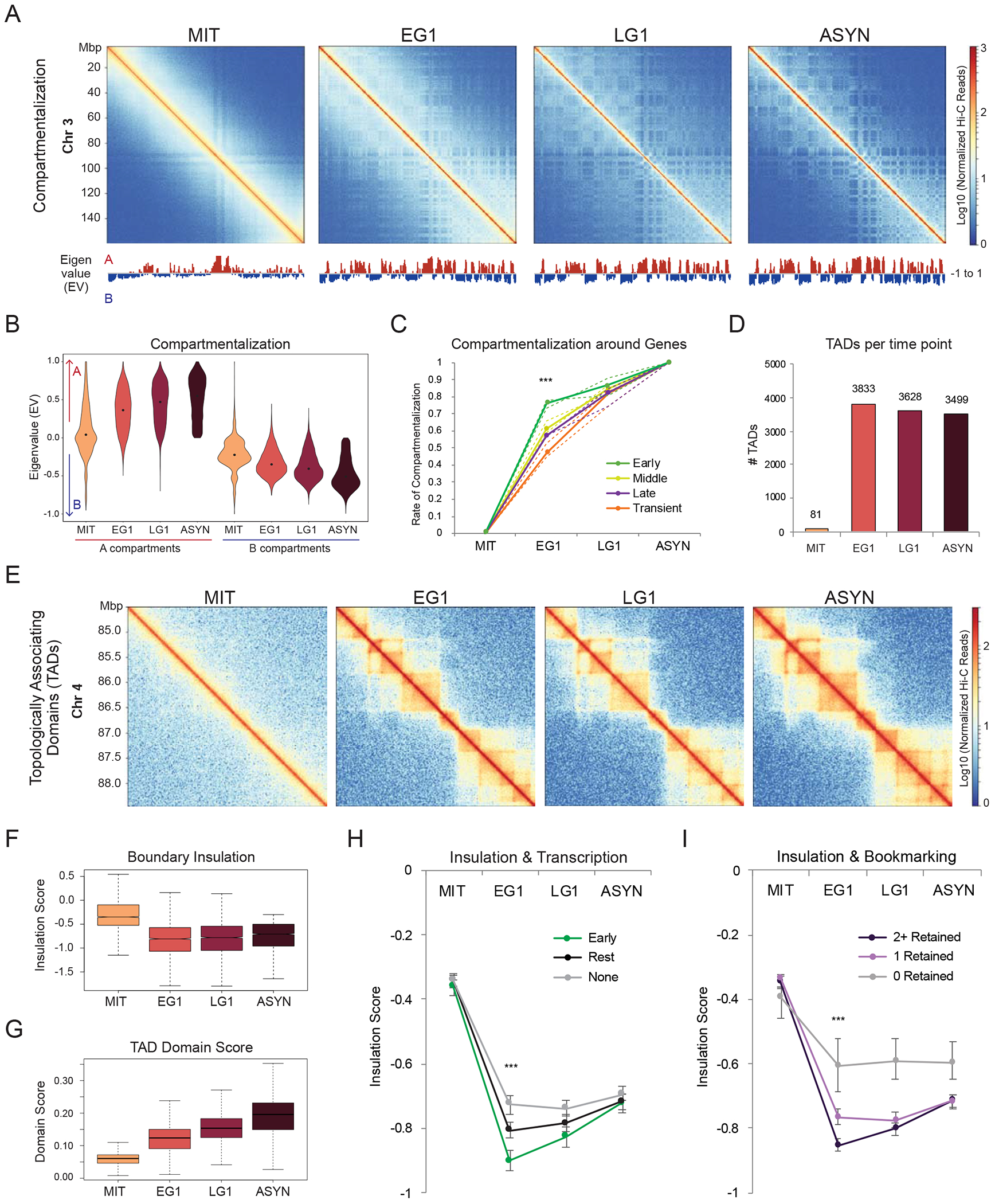
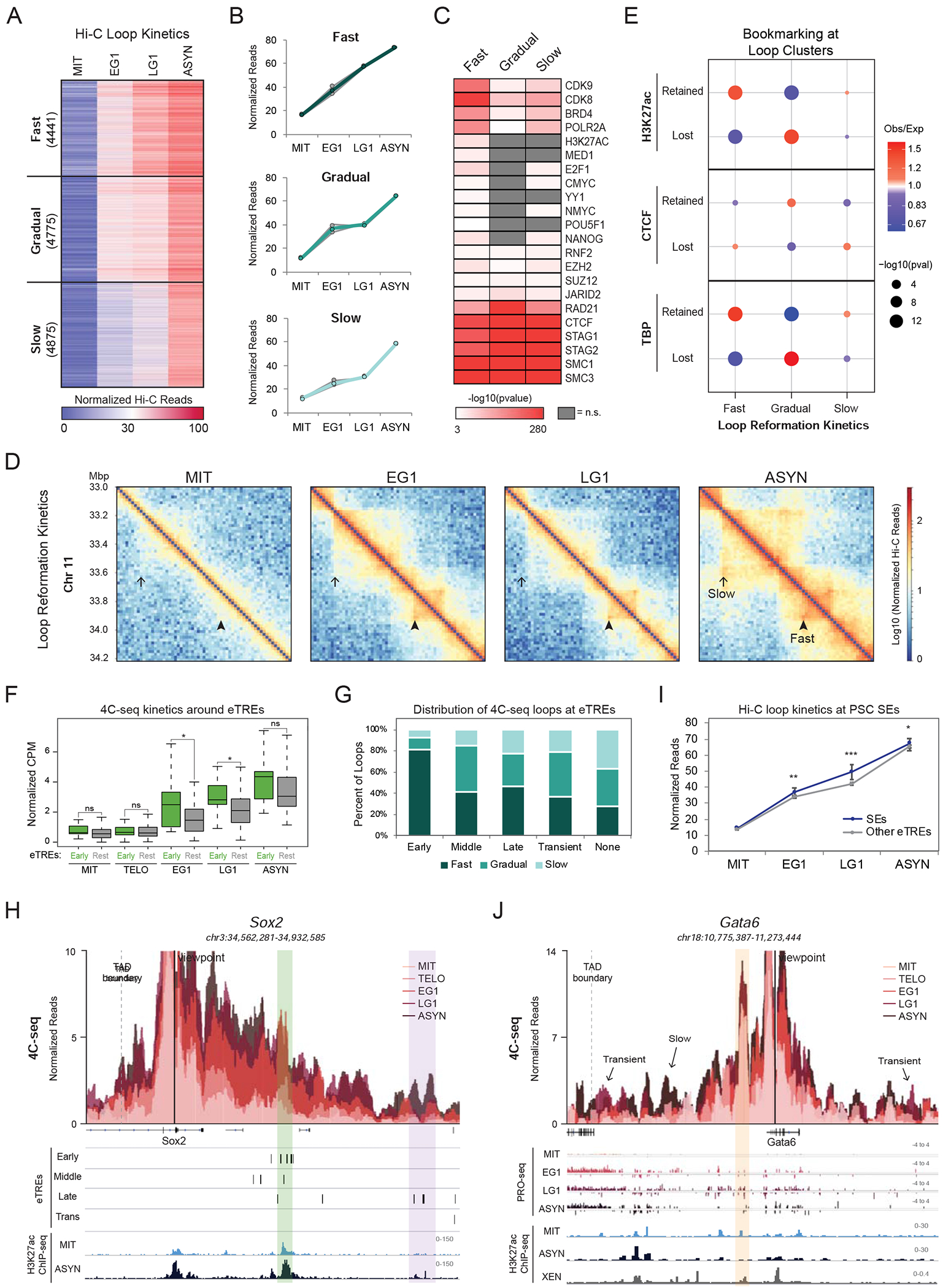
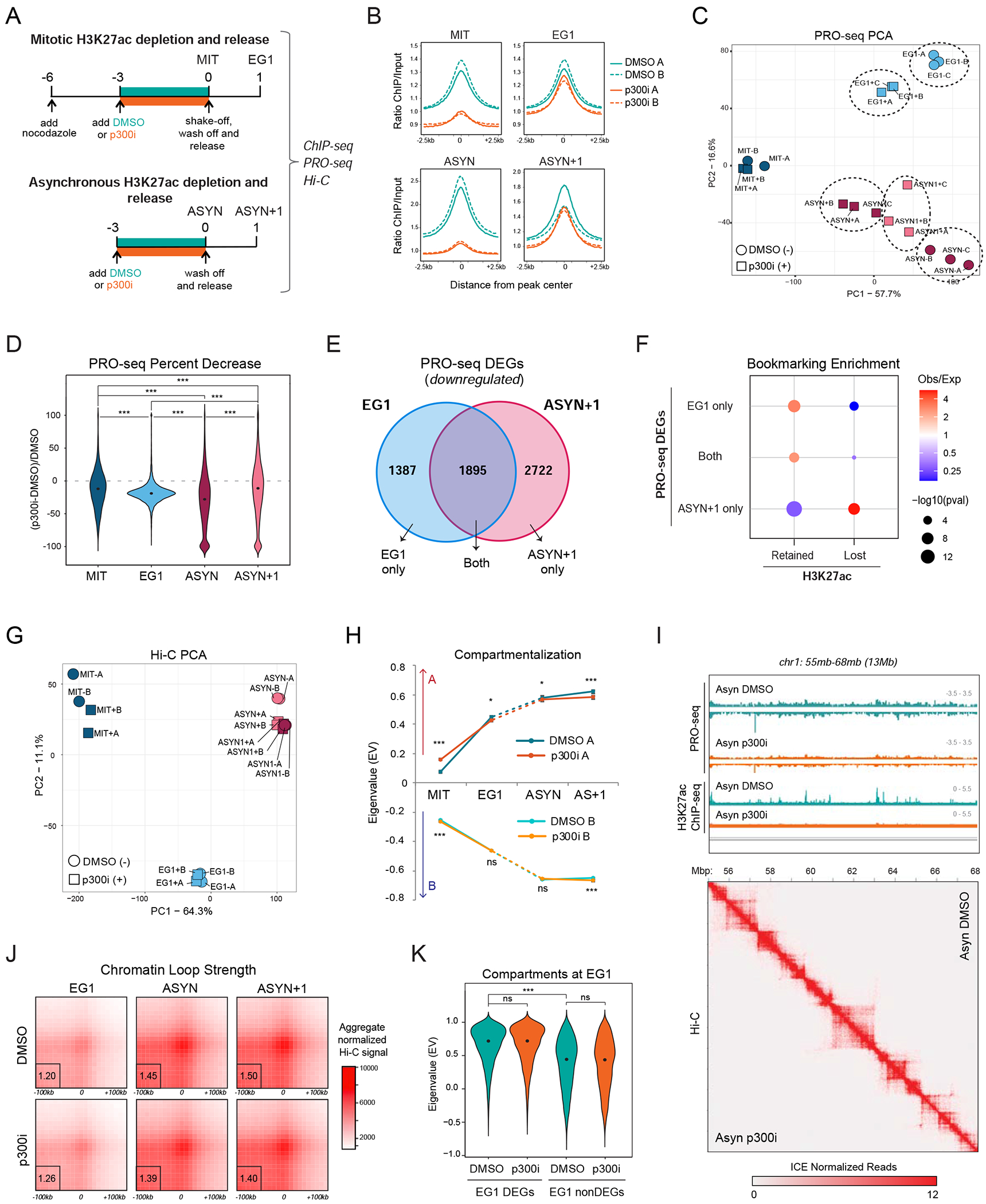
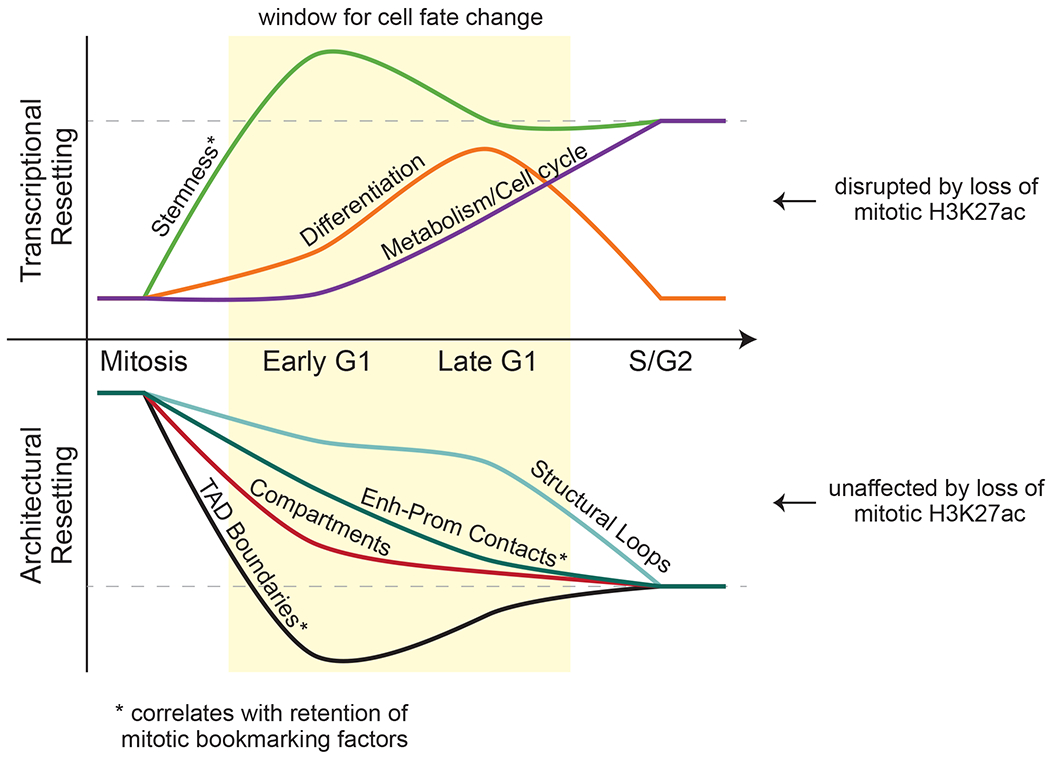
During self-renewal, cell-type-defining features are drastically perturbed in mitosis and must be faithfully reestablished upon G1 entry, a process that remains largely elusive. Here, we characterized at a genome-wide scale the dynamic transcriptional and architectural resetting of mouse pluripotent stem cells (PSCs) upon mitotic exit. We captured distinct waves of transcriptional reactivation with rapid induction of stem cell genes and transient activation of lineage-specific genes. Topological reorganization at different hierarchical levels also occurred in an asynchronous manner and showed partial coordination with transcriptional resetting. Globally, rapid transcriptional and architectural resetting associated with mitotic retention of H3K27 acetylation, supporting a bookmarking function. Indeed, mitotic depletion of H3K27ac impaired the early reactivation of bookmarked, stem-cell-associated genes. However, 3D chromatin reorganization remained largely unaffected, suggesting that these processes are driven by distinct forces upon mitotic exit. This study uncovers principles and mediators of PSC molecular resetting during self-renewal.
Keywords: 3D chromatin organization; H3K27ac; Hi-C; PRO-seq; bookmarking; cell identity; enhancer-promoter interaction; mitosis; pluripotent stem cells; transcription reactivation.
Copyright © 2021 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
