

Targeting FAK in anticancer combination therapies
- PMID: 33731845
- PMCID: PMC8276817
- DOI: 10.1038/s41568-021-00340-6
Targeting FAK in anticancer combination therapies
Abstract
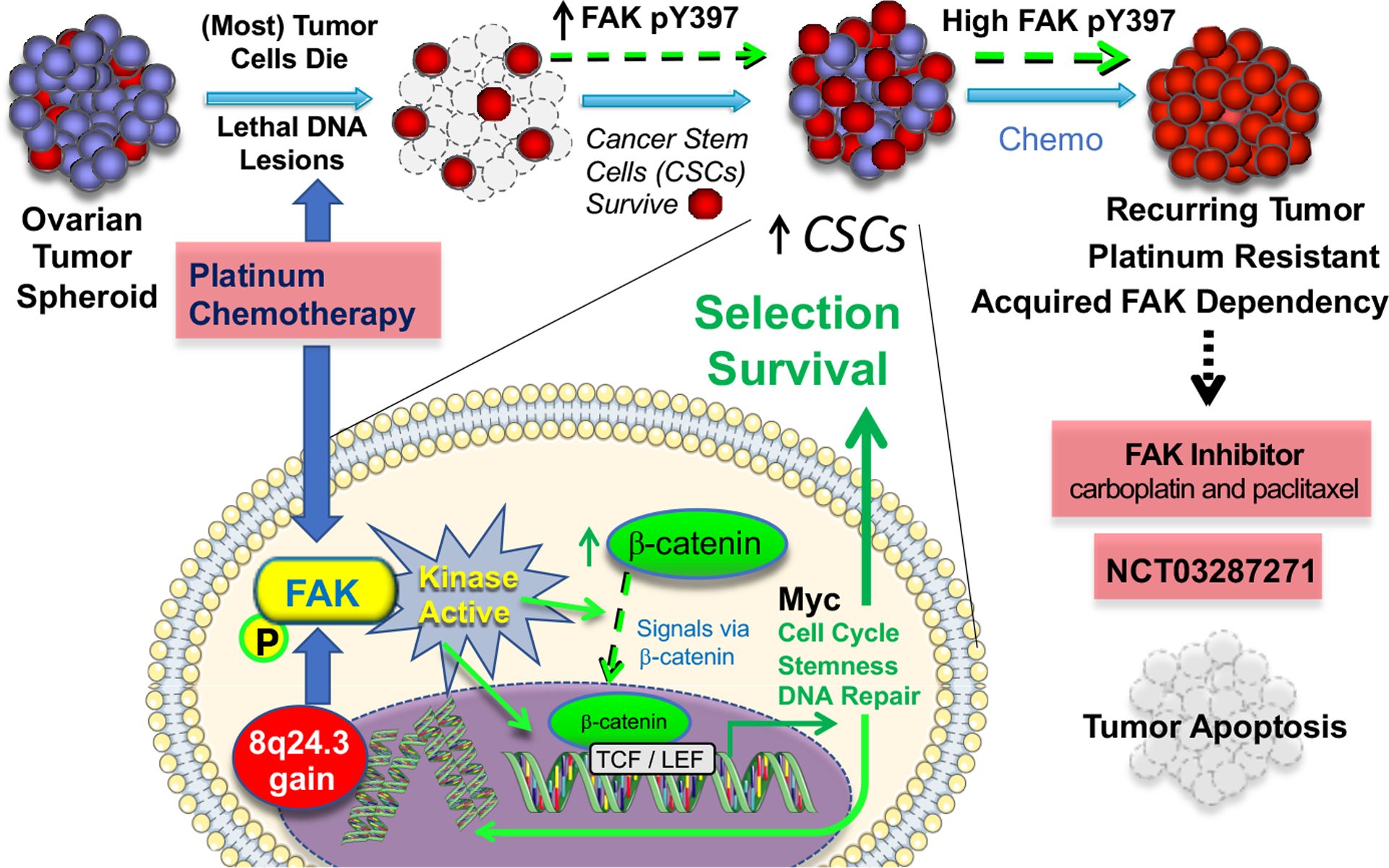
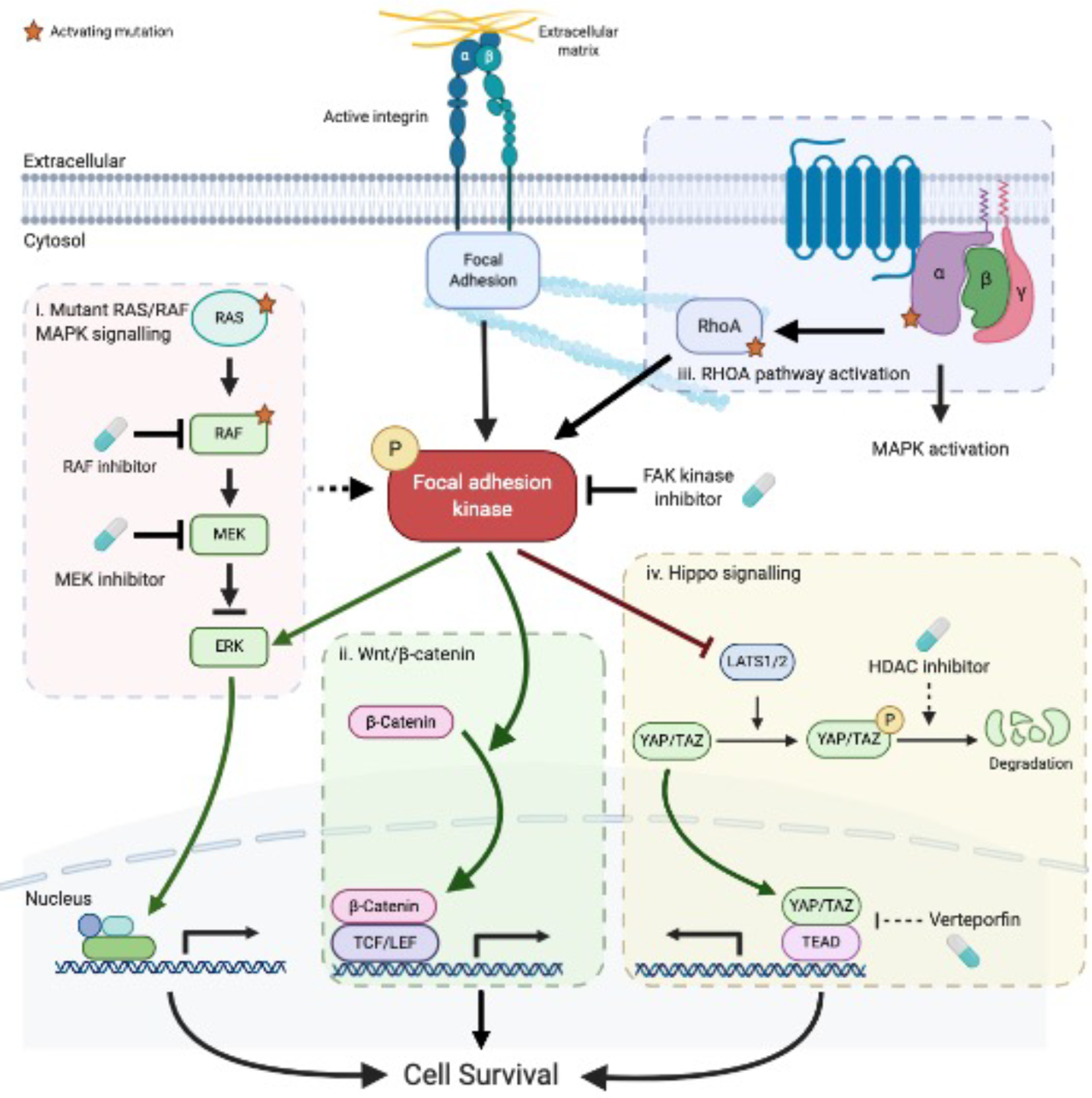
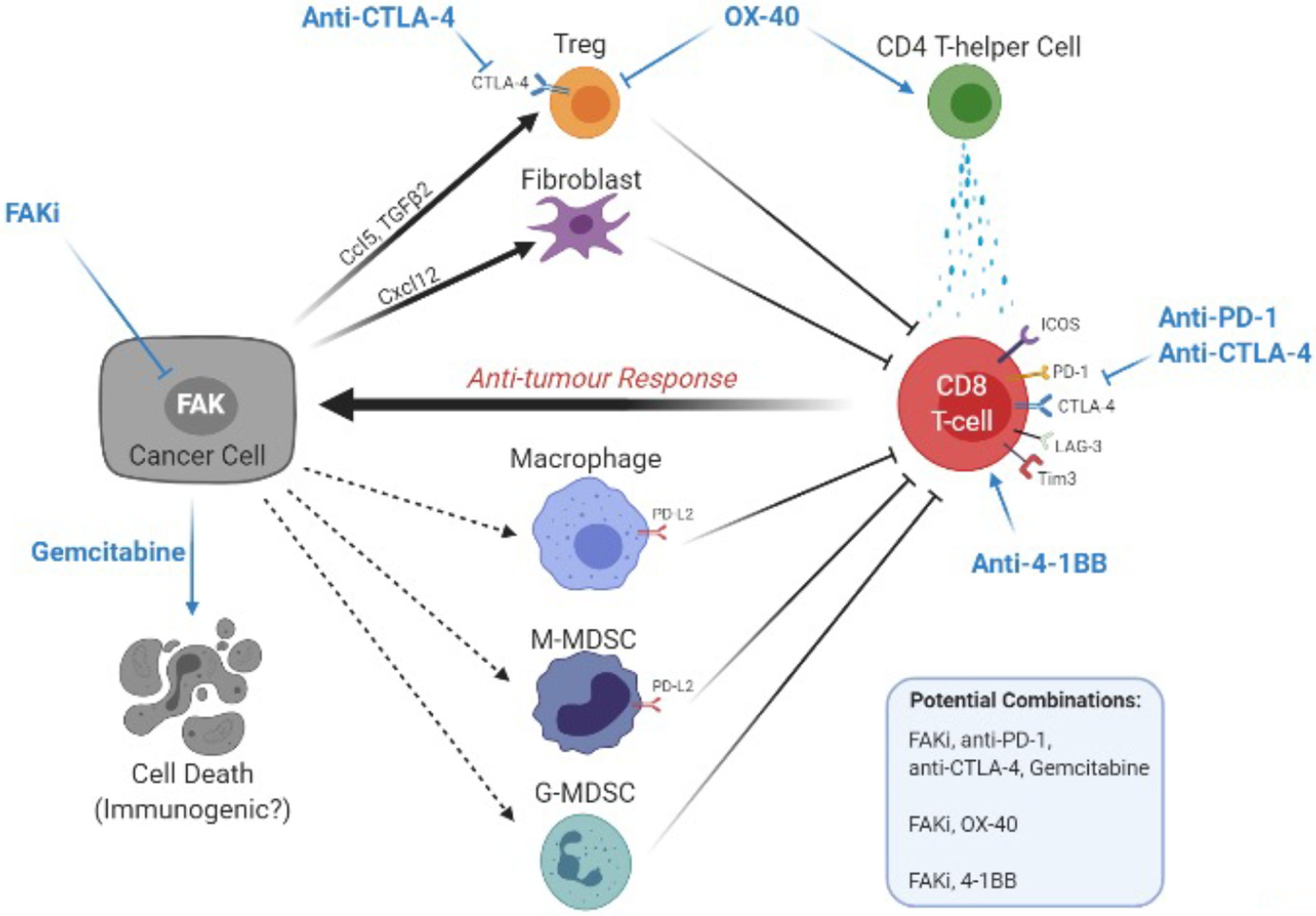
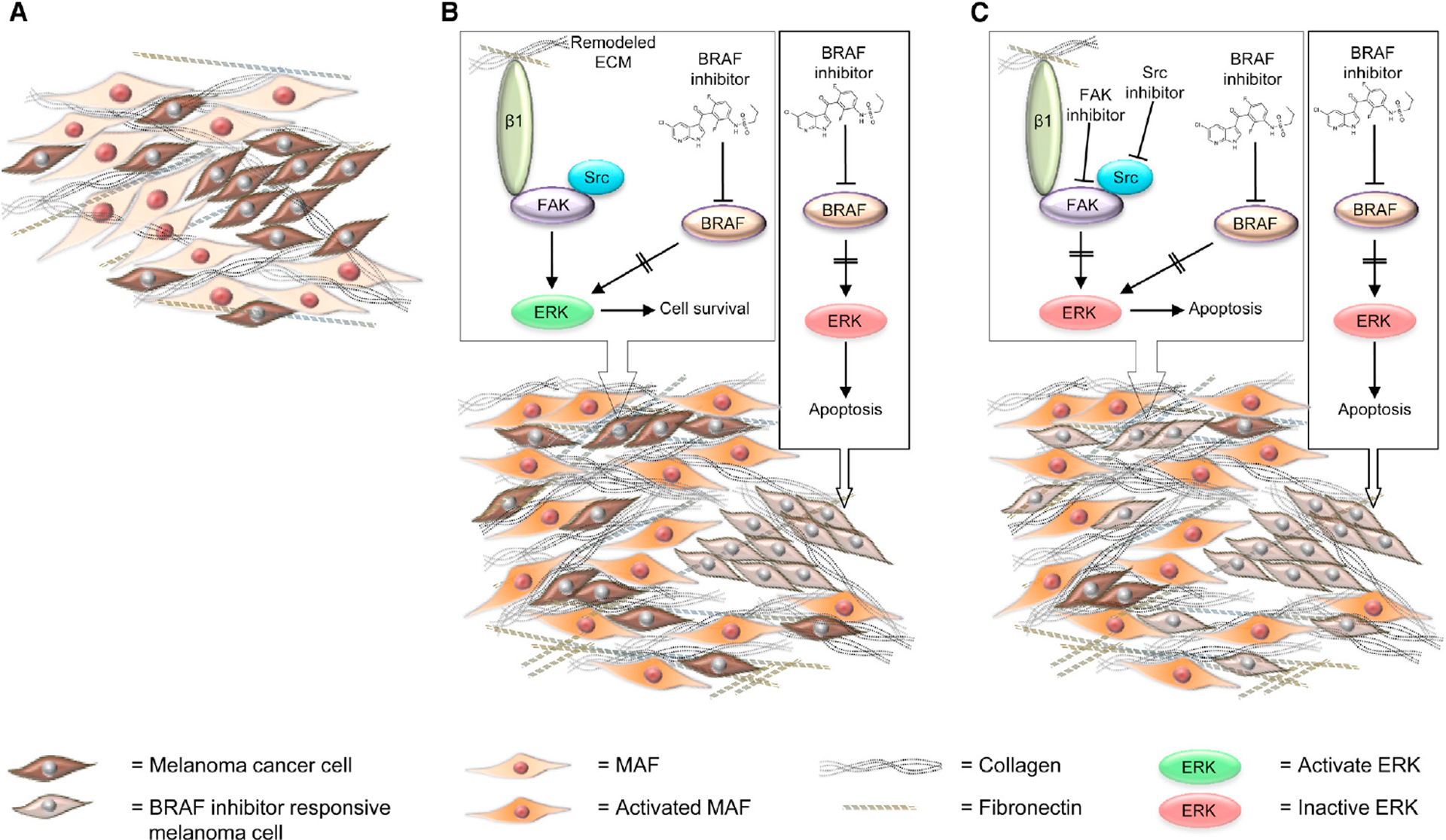
Focal adhesion kinase (FAK) is both a non-receptor tyrosine kinase and an adaptor protein that primarily regulates adhesion signalling and cell migration, but FAK can also promote cell survival in response to stress. FAK is commonly overexpressed in cancer and is considered a high-value druggable target, with multiple FAK inhibitors currently in development. Evidence suggests that in the clinical setting, FAK targeting will be most effective in combination with other agents so as to reverse failure of chemotherapies or targeted therapies and enhance efficacy of immune-based treatments of solid tumours. Here, we discuss the recent preclinical evidence that implicates FAK in anticancer therapeutic resistance, leading to the view that FAK inhibitors will have their greatest utility as combination therapies in selected patient populations.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
