

A Phase 3 Open-Label Study of Elexacaftor/Tezacaftor/Ivacaftor in Children 6 through 11 Years of Age with Cystic Fibrosis and at Least One F508del Allele
- PMID: 33734030
- PMCID: PMC8483230
- DOI: 10.1164/rccm.202102-0509OC
A Phase 3 Open-Label Study of Elexacaftor/Tezacaftor/Ivacaftor in Children 6 through 11 Years of Age with Cystic Fibrosis and at Least One F508del Allele
Abstract
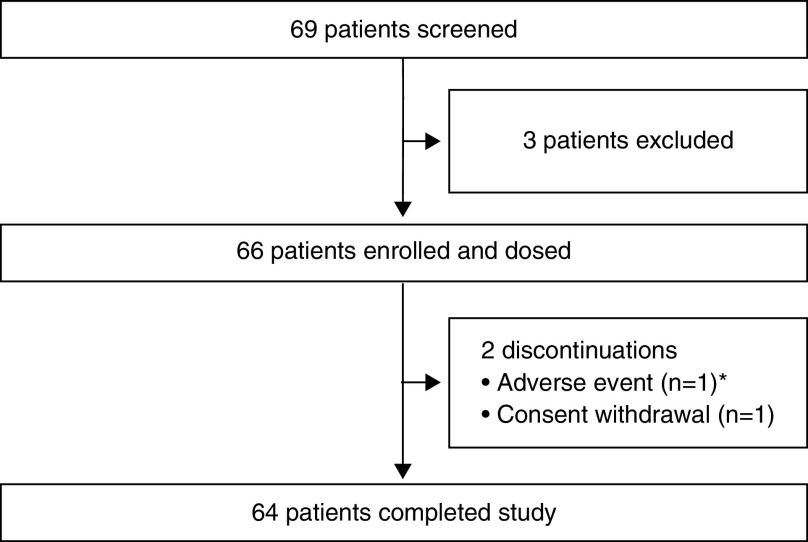
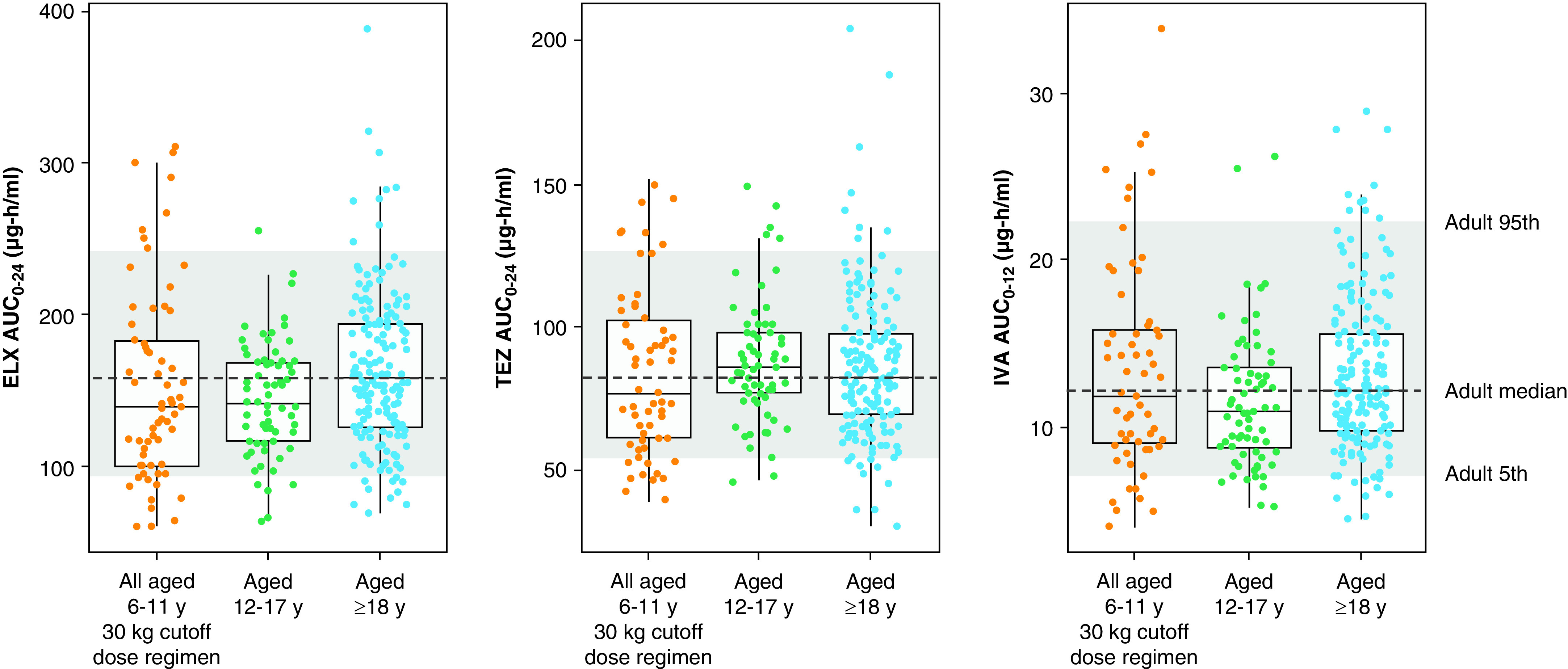
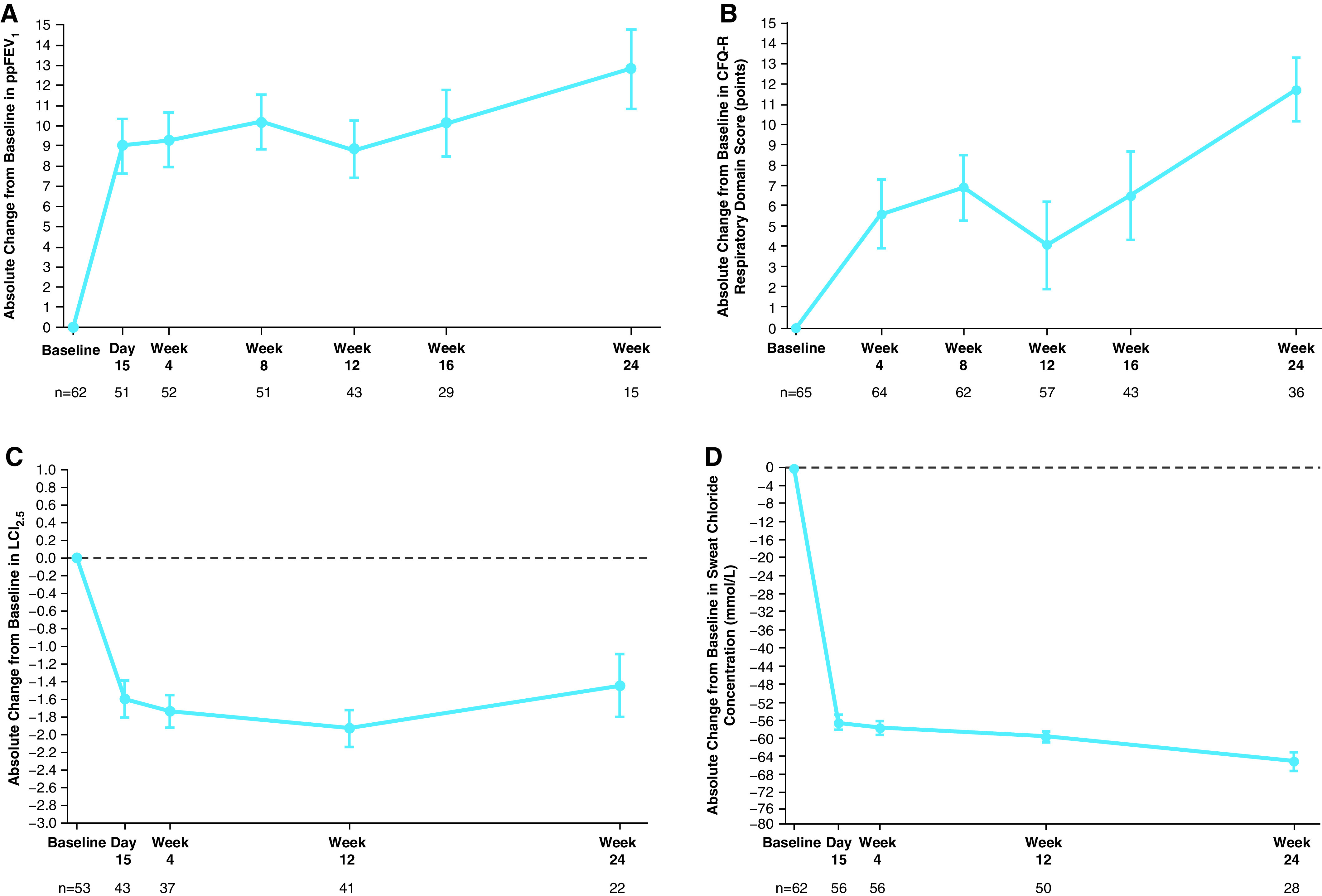
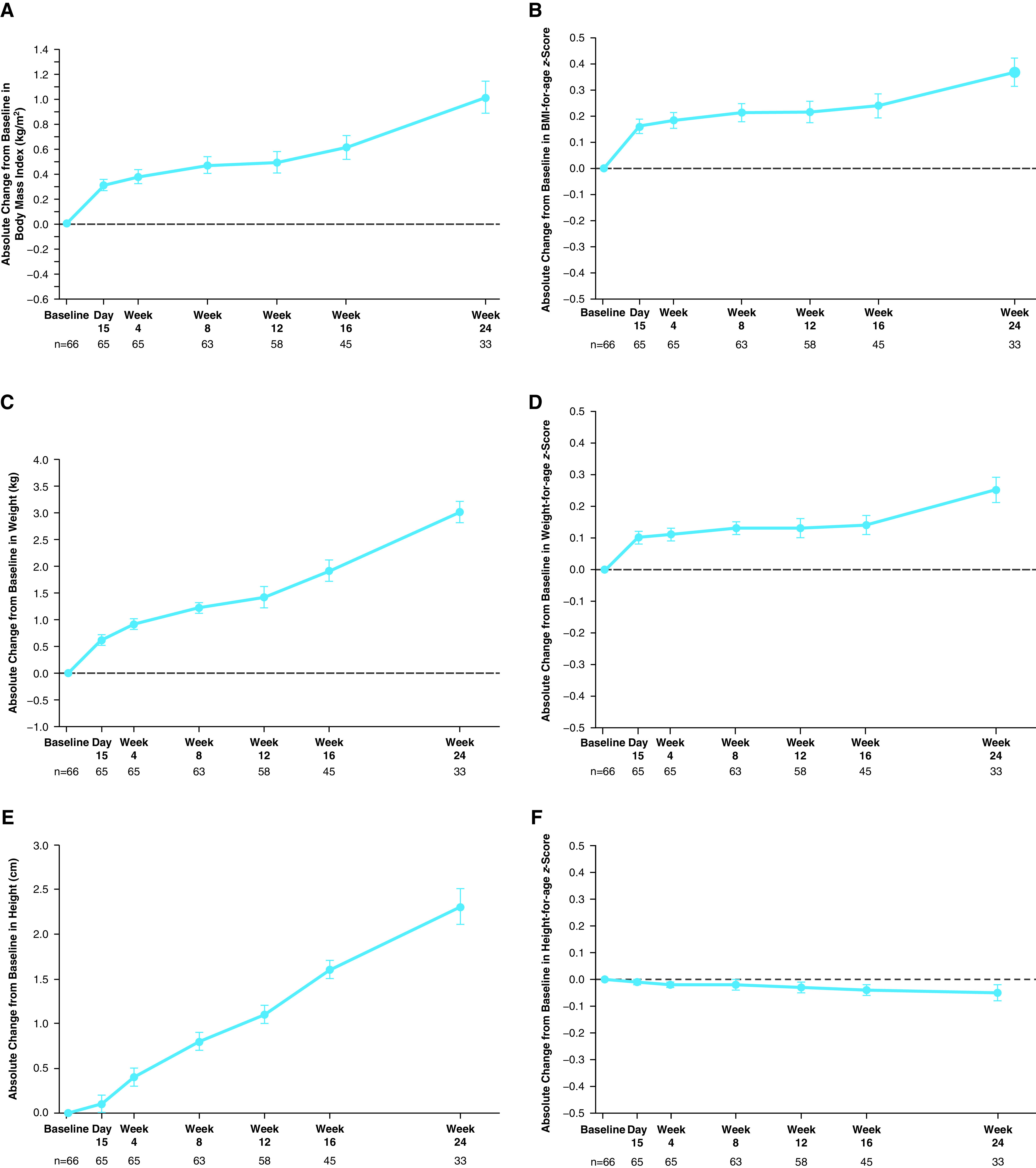
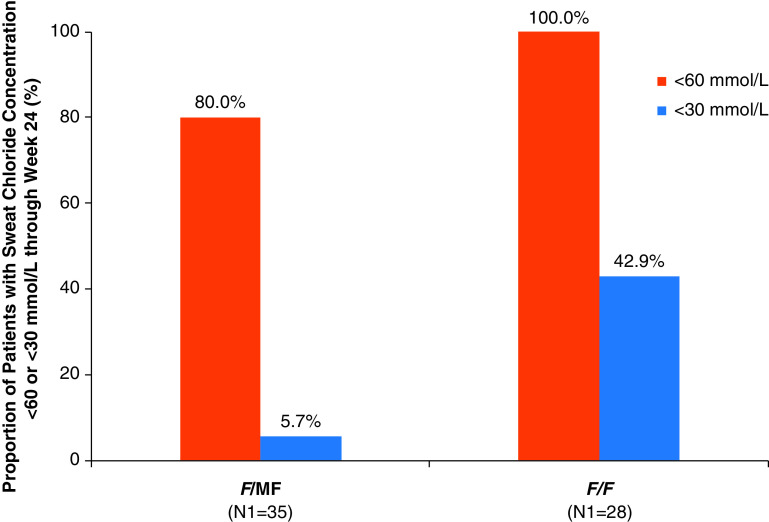
Rationale: Elexacaftor/tezacaftor/ivacaftor (ELX/TEZ/IVA) was shown to be efficacious and safe in patients ≥12 years of age with cystic fibrosis and at least one F508del-CFTR (cystic fibrosis transmembrane conductance regulator) allele, but it has not been evaluated in children <12 years of age. Objectives: To assess the safety, pharmacokinetics, and efficacy of ELX/TEZ/IVA in children 6 through 11 years of age with F508del-minimal function or F508del-F508del genotypes. Methods: In this 24-week open-label phase 3 study, children (N = 66) weighing <30 kg received 50% of the ELX/TEZ/IVA adult daily dose (ELX 100 mg once daily, TEZ 50 mg once daily, and IVA 75 mg every 12 h) whereas children weighing ⩾30 kg received the full adult daily dose (ELX 200 mg once daily, TEZ 100 mg once daily, and IVA 150 mg every 12 h). Measurements and Main Results: The primary endpoint was safety and tolerability. The safety and pharmacokinetic profiles of ELX/TEZ/IVA were generally consistent with those observed in older patients. The most commonly reported adverse events included cough, headache, and pyrexia; in most of the children who had adverse events, these were mild or moderate in severity. Through Week 24, ELX/TEZ/IVA treatment improved the percentage of predicted FEV1 (10.2 percentage points; 95% confidence interval [CI], 7.9 to 12.6), Cystic Fibrosis Questionnaire-Revised respiratory domain score (7.0 points; 95% CI, 4.7 to 9.2), lung clearance index2.5 (-1.71 units; 95% CI, -2.11 to -1.30), and sweat chloride (-60.9 mmol/L; 95% CI, -63.7 to -58.2); body mass index-for-age z-score increased over the 24-week treatment period when compared with the pretreatment baseline. Conclusions: Our results show ELX/TEZ/IVA is safe and efficacious in children 6 through 11 years of age with at least one F508del-CFTR allele, supporting its use in this patient population. Clinical trial registered with www.clinicaltrials.gov (NCT03691779).
Keywords: child; cystic fibrosis; elexacaftor; ivacaftor; tezacaftor.
Figures
Comment in
-
The Future of Highly Effective Modulator Therapy in Cystic Fibrosis.Am J Respir Crit Care Med. 2021 Jun 15;203(12):1453-1455. doi: 10.1164/rccm.202104-0850ED. Am J Respir Crit Care Med. 2021. PMID: 33901406 Free PMC article. No abstract available.
References
-
- Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. - PubMed
-
- Cystic Fibrosis Foundation. Bethesda, MD: Cystic Fibrosis Foundation; 2020. https://www.cff.org/Research/Researcher-Resources/Patient-Registry/2019-...
-
- Elborn JS. Cystic fibrosis. Lancet. 2016;388:2519–2531. - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
