

Whole-organism eQTL mapping at cellular resolution with single-cell sequencing
- PMID: 33734084
- PMCID: PMC8062134
- DOI: 10.7554/eLife.65857
Whole-organism eQTL mapping at cellular resolution with single-cell sequencing
Abstract
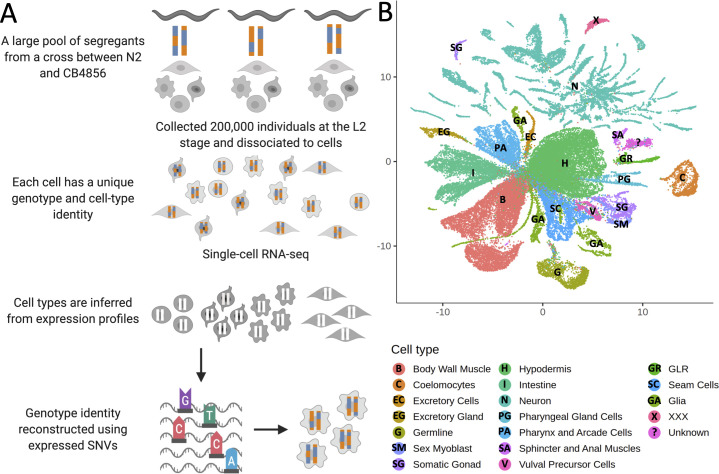
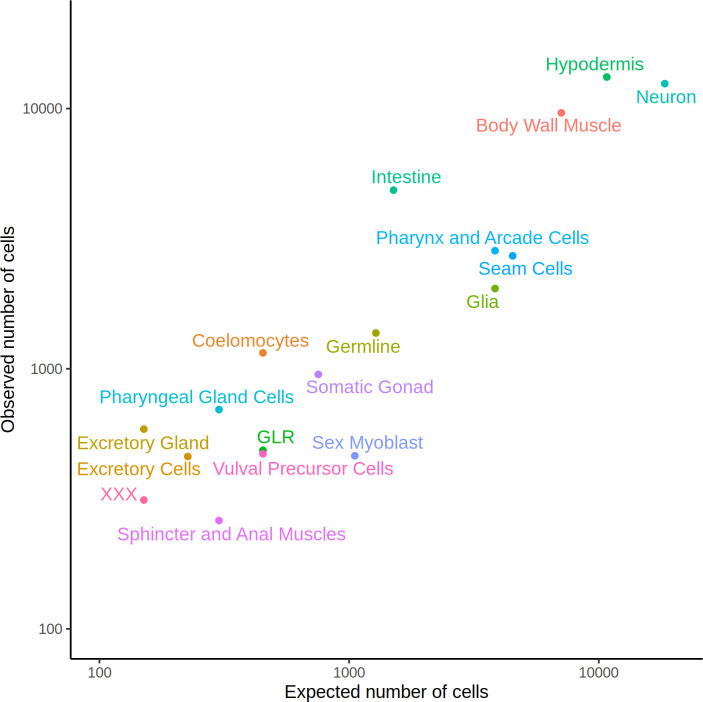
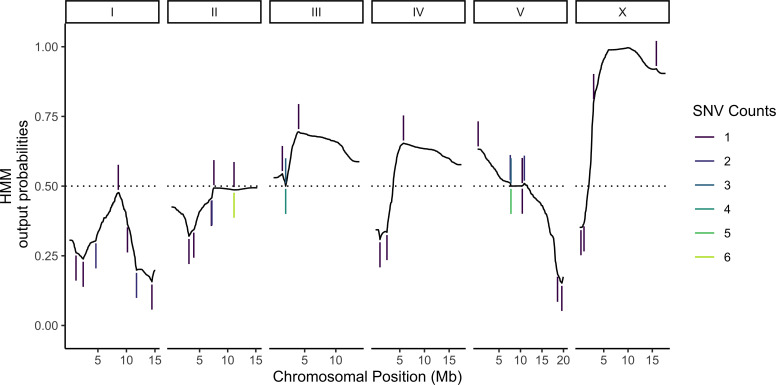
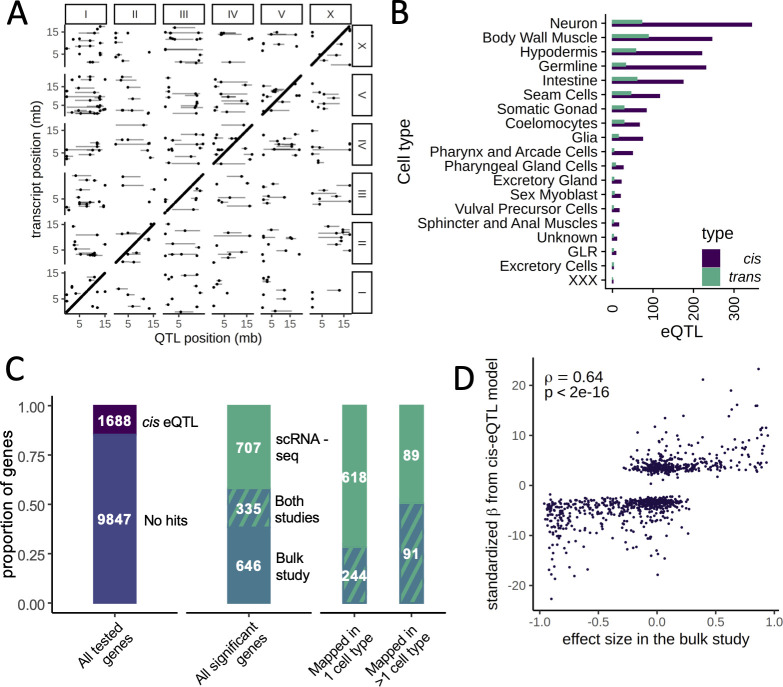
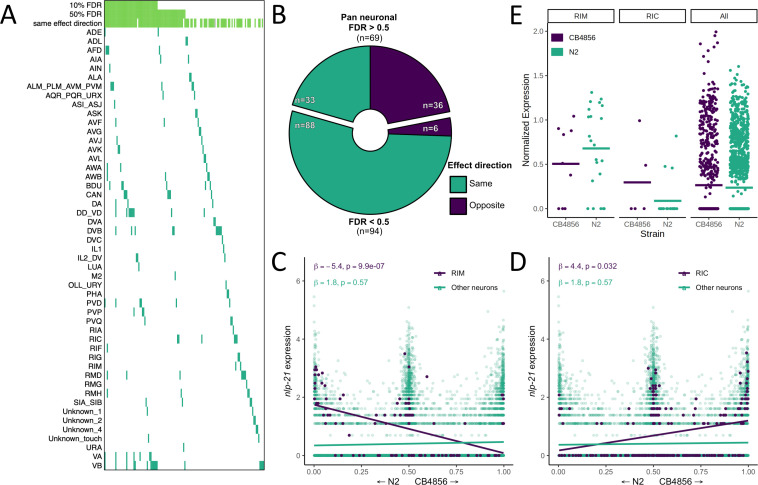
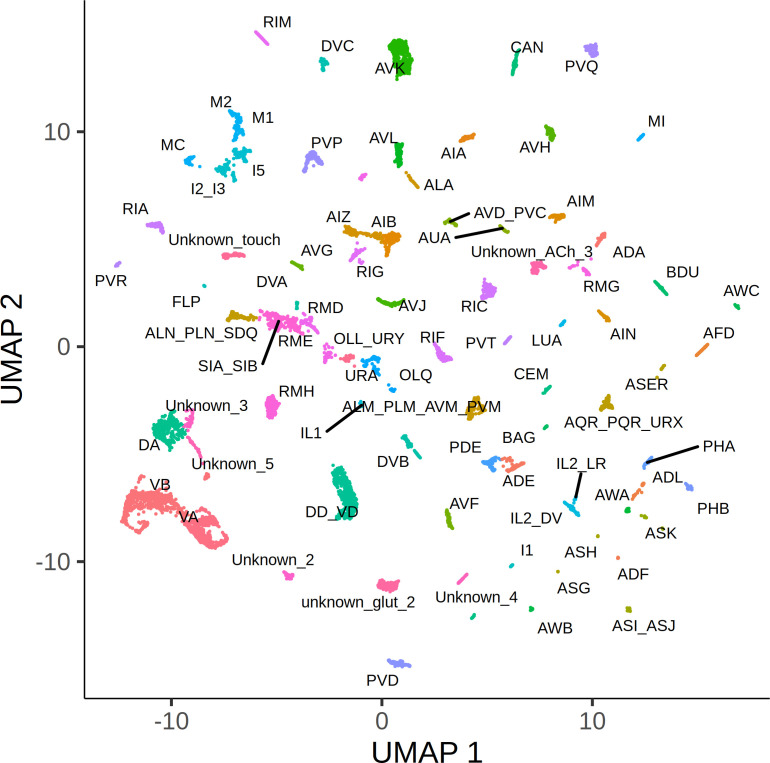
Genetic regulation of gene expression underlies variation in disease risk and other complex traits. The effect of expression quantitative trait loci (eQTLs) varies across cell types; however, the complexity of mammalian tissues makes studying cell-type eQTLs highly challenging. We developed a novel approach in the model nematode Caenorhabditis elegans that uses single-cell RNA sequencing to map eQTLs at cellular resolution in a single one-pot experiment. We mapped eQTLs across cell types in an extremely large population of genetically distinct C. elegans individuals. We found cell-type-specific trans eQTL hotspots that affect the expression of core pathways in the relevant cell types. Finally, we found single-cell-specific eQTL effects in the nervous system, including an eQTL with opposite effects in two individual neurons. Our results show that eQTL effects can be specific down to the level of single cells.
Keywords: C. elegans; cell-types; eqtl; genetics; genomics; mapping; rna-seq; single-cell.
Plain language summary
DNA sequences that differ between individuals often change the activation pattern of genes. That is, they change how, when, or why genes switch on and off. We call such DNA sequences 'expression quantitative trait loci', or eQTLs for short. Many of these eQTLs affect biological traits, but their effects are not always easy to predict. In fact, these effects can change with time, with different stress levels, and even in different types of cells. This makes studying eQTLs challenging, especially in organisms with many different types of cells. Standard methods of studying eQTLs involve separately measuring which genes switch on or off under every condition and in each cell. However, a technology called single cell sequencing makes it possible to profile many cells simultaneously, determining which genes are switched on or off in each one. Applying this technology to eQTL discovery could make a challenging problem solvable with a straightforward experiment. To test this idea, Ben-David et al. worked with the nematode worm Caenorhabditis elegans, a frequently-used experimental animal which has a small number of cells with well-defined types. The experiment included tens of thousands of cells from tens of thousands of genetically distinct worms. Using single cell sequencing, Ben-David et al. were able to find eQTLs across all the different cell types in the worms. These included many eQTLs already identified using traditional approaches, confirming that the new method worked. To understand the effects of some of these eQTLs in more detail, Ben-David et al. took a closer look at the cells of the nervous system. This revealed that eQTL effects not only differ between cell types, but also between individual cells. In one example, an eQTL changed the expression of the same gene in opposite directions in two different nerve cells. There is great interest in studying eQTLs because they provide a common mechanism by which changes in DNA can affect biological traits, including diseases. These experiments highlight the need to compare eQTLs in all conditions and tissues of interest, and the new technique provides a simpler way to do so. As single-cell technology matures and enables profiling of larger numbers of cells, it should become possible to analyze more complex organisms. This could one day include mammals.
© 2021, Ben-David et al.
Conflict of interest statement
EB, JB, LG, SZ, JB, LK No competing interests declared
Figures
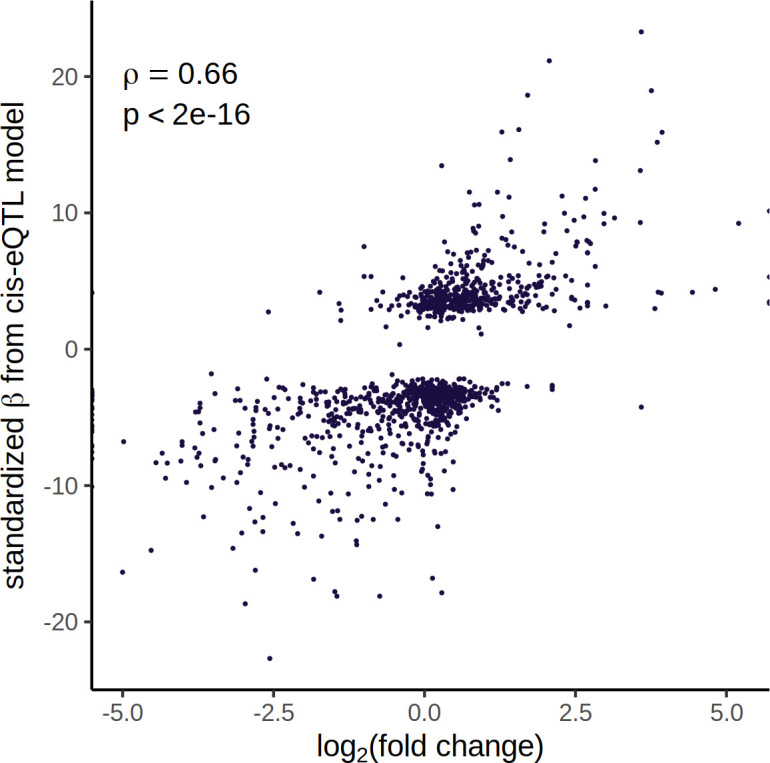
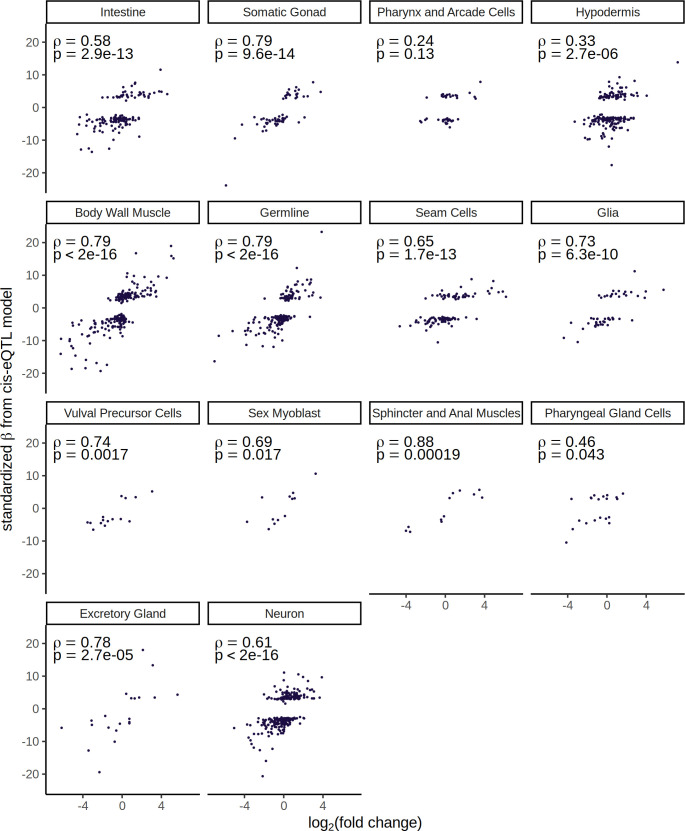
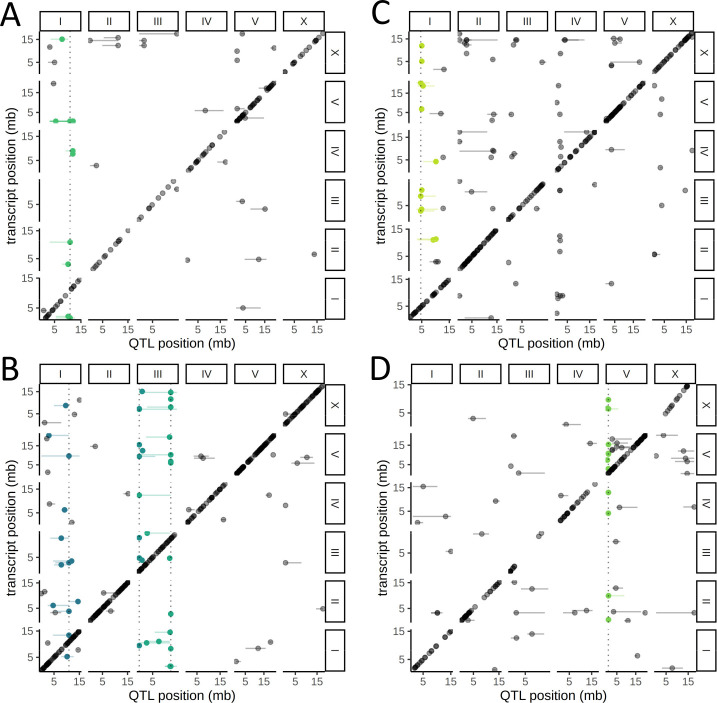
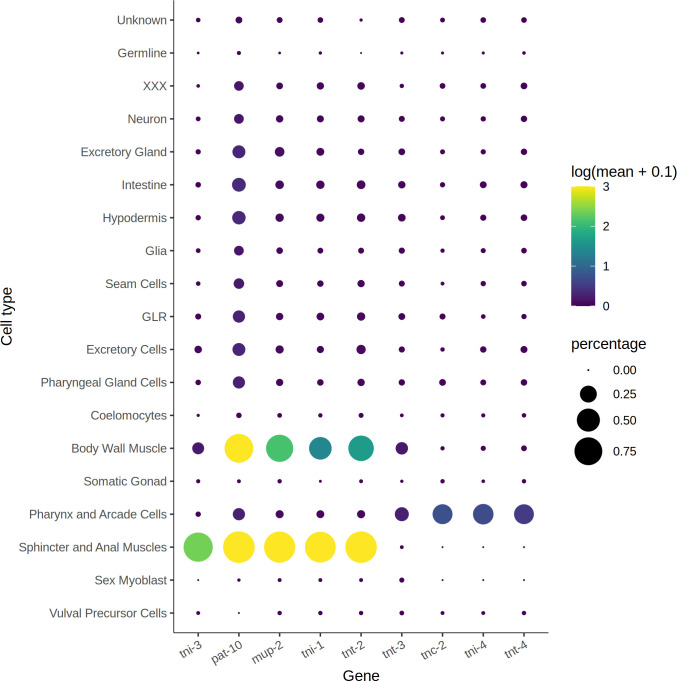
References
-
- Andersen EC, Shimko TC, Crissman JR, Ghosh R, Bloom JS, Seidel HS, Gerke JP, Kruglyak L. A Powerful New Quantitative Genetics Platform, Combining Caenorhabditis elegans High-Throughput Fitness Assays with a Large Collection of Recombinant Strains. G3: Genes, Genomes, Genetics. 2015;5:911–920. doi: 10.1534/g3.115.017178. - DOI - PMC - PubMed
Publication types
MeSH terms
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
