

Epigenetic inactivation of ERF reactivates γ-globin expression in β-thalassemia
- PMID: 33735615
- PMCID: PMC8059375
- DOI: 10.1016/j.ajhg.2021.03.005
Epigenetic inactivation of ERF reactivates γ-globin expression in β-thalassemia
Abstract
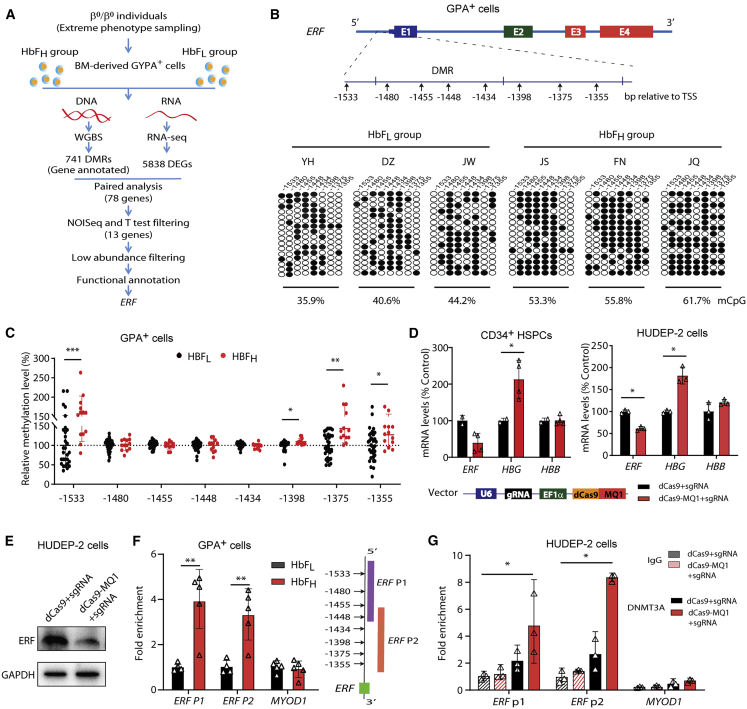
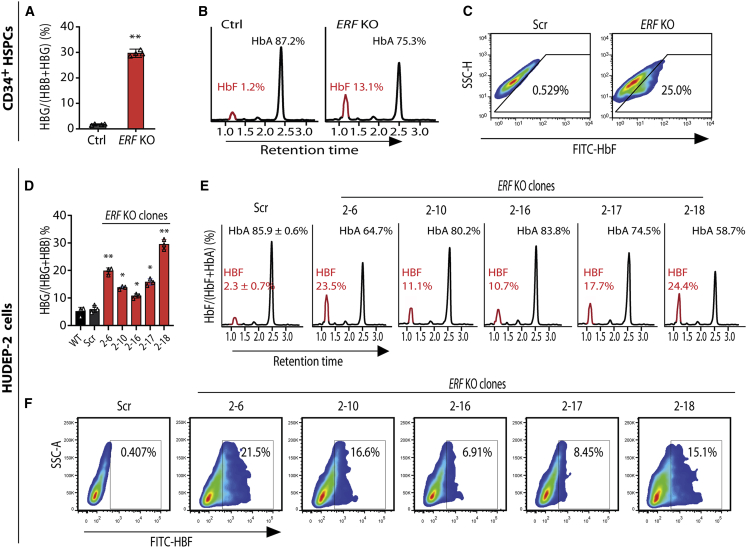
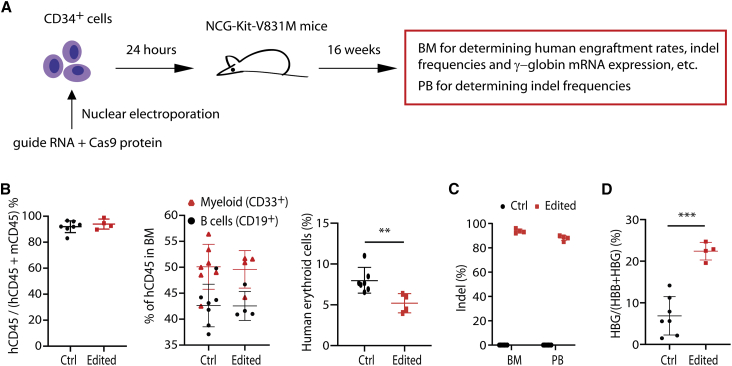
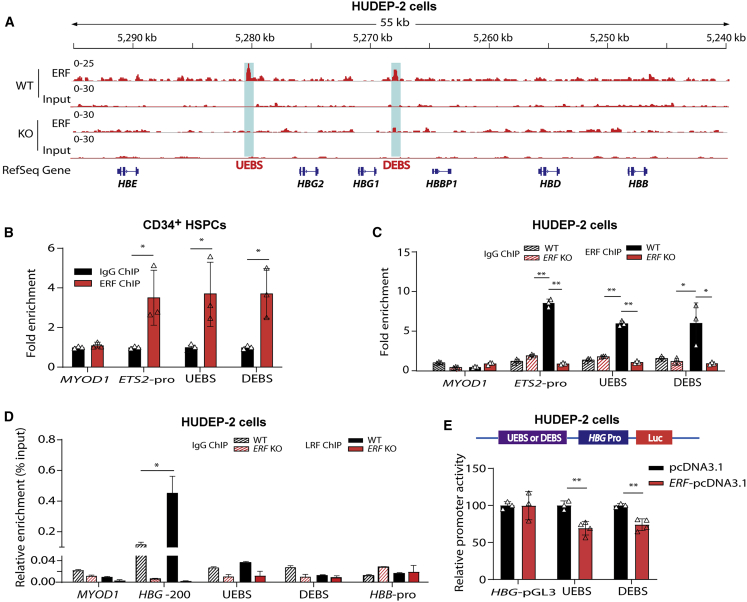
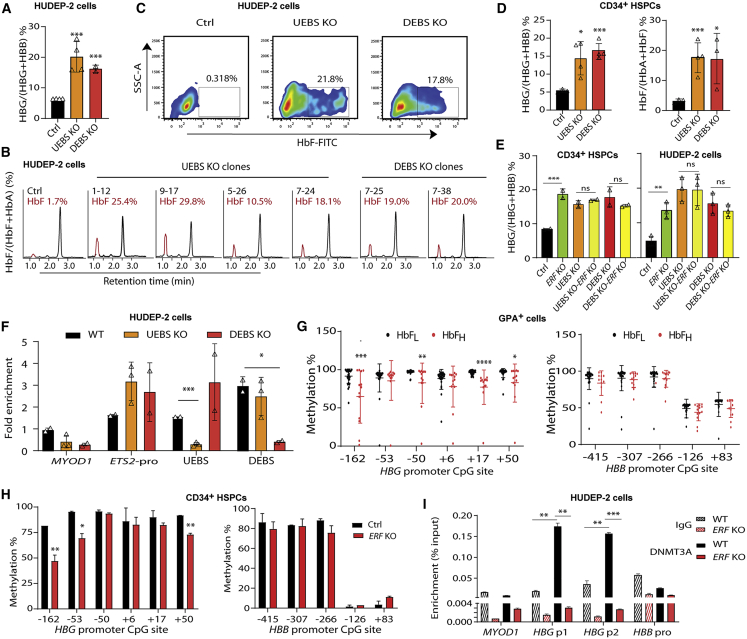
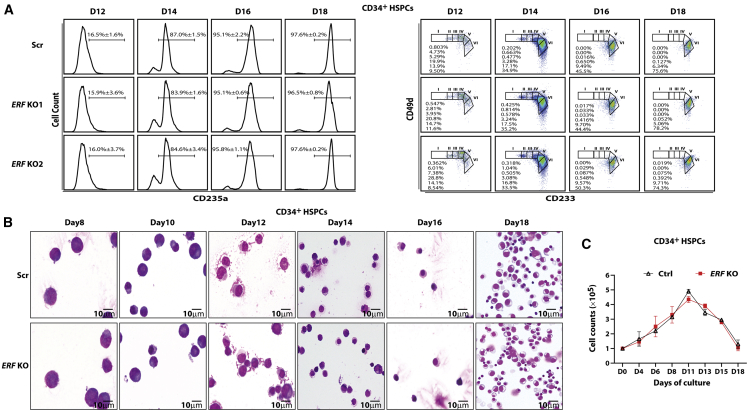
The fetal-to-adult hemoglobin switch is regulated in a developmental stage-specific manner and reactivation of fetal hemoglobin (HbF) has therapeutic implications for treatment of β-thalassemia and sickle cell anemia, two major global health problems. Although significant progress has been made in our understanding of the molecular mechanism of the fetal-to-adult hemoglobin switch, the mechanism of epigenetic regulation of HbF silencing remains to be fully defined. Here, we performed whole-genome bisulfite sequencing and RNA sequencing analysis of the bone marrow-derived GYPA+ erythroid cells from β-thalassemia-affected individuals with widely varying levels of HbF groups (HbF ≥ 95th percentile or HbF ≤ 5th percentile) to screen epigenetic modulators of HbF and phenotypic diversity of β-thalassemia. We identified an ETS2 repressor factor encoded by ERF, whose promoter hypermethylation and mRNA downregulation are associated with high HbF levels in β-thalassemia. We further observed that hypermethylation of the ERF promoter mediated by enrichment of DNMT3A leads to demethylation of γ-globin genes and attenuation of binding of ERF on the HBG promoter and eventually re-activation of HbF in β-thalassemia. We demonstrated that ERF depletion markedly increased HbF production in human CD34+ erythroid progenitor cells, HUDEP-2 cell lines, and transplanted NCG-Kit-V831M mice. ERF represses γ-globin expression by directly binding to two consensus motifs regulating γ-globin gene expression. Importantly, ERF depletion did not affect maturation of erythroid cells. Identification of alterations in DNA methylation of ERF as a modulator of HbF synthesis opens up therapeutic targets for β-hemoglobinopathies.
Keywords: CD34+ HSPCs; ERF; GYPA+ cells; engraftment mice; epigenetics; fetal hemoglobin; genome editing; methylation; whole-genome bisulfite sequencing; β-thalassemia.
Copyright © 2021. Published by Elsevier Inc.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Vinjamur D.S., Bauer D.E., Orkin S.H. Recent progress in understanding and manipulating haemoglobin switching for the haemoglobinopathies. Br. J. Haematol. 2018;180:630–643. - PubMed
-
- Wienert B., Martyn G.E., Funnell A.P.W., Quinlan K.G.R., Crossley M. Wake-up Sleepy Gene: Reactivating Fetal Globin for β-Hemoglobinopathies. Trends Genet. 2018;34:927–940. - PubMed
-
- Mettananda S., Higgs D.R. Molecular Basis and Genetic Modifiers of Thalassemia. Hematol. Oncol. Clin. North Am. 2018;32:177–191. - PubMed
-
- Chondrou V., Kolovos P., Sgourou A., Kourakli A., Pavlidaki A., Kastrinou V., John A., Symeonidis A., Ali B.R., Papachatzopoulou A. Whole transcriptome analysis of human erythropoietic cells during ontogenesis suggests a role of VEGFA gene as modulator of fetal hemoglobin and pharmacogenomic biomarker of treatment response to hydroxyurea in β-type hemoglobinopathy patients. Hum. Genomics. 2017;11:24. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
