

Fine-tuning the performance of ddRAD-seq in the peach genome
- PMID: 33737671
- PMCID: PMC7973760
- DOI: 10.1038/s41598-021-85815-0
Fine-tuning the performance of ddRAD-seq in the peach genome
Abstract
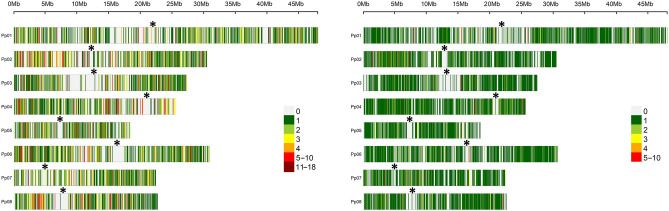
The advance of Next Generation Sequencing (NGS) technologies allows high-throughput genotyping at a reasonable cost, although, in the case of peach, this technology has been scarcely developed. To date, only a standard Genotyping by Sequencing approach (GBS), based on a single restriction with ApeKI to reduce genome complexity, has been applied in peach. In this work, we assessed the performance of the double-digest RADseq approach (ddRADseq), by testing 6 double restrictions with the restriction profile generated with ApeKI. The enzyme pair PstI/MboI retained the highest number of loci in concordance with the in silico analysis. Under this condition, the analysis of a diverse germplasm collection (191 peach genotypes) yielded 200,759,000 paired-end (2 × 250 bp) reads that allowed the identification of 113,411 SNP, 13,661 InDel and 2133 SSR. We take advantage of a wide sample set to describe technical scope of the platform. The novel platform presented here represents a useful tool for genomic-based breeding for peach.
Conflict of interest statement
The authors declare no competing interests.
Figures

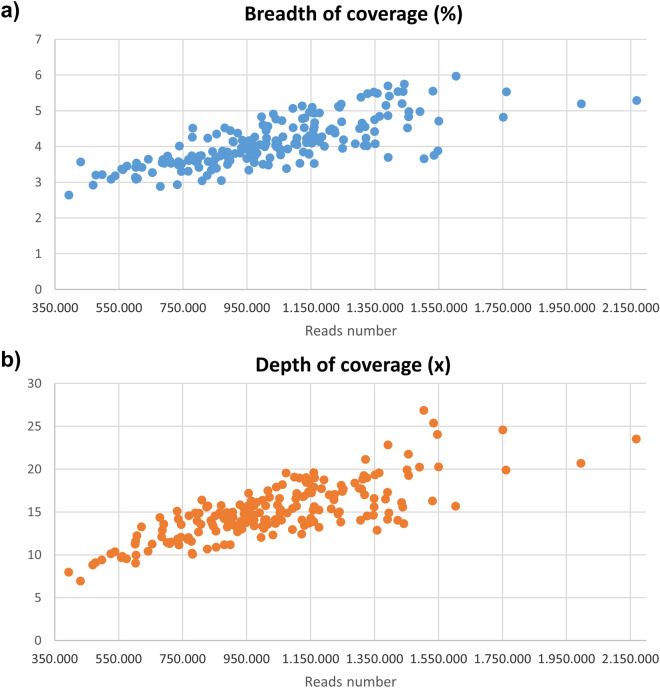

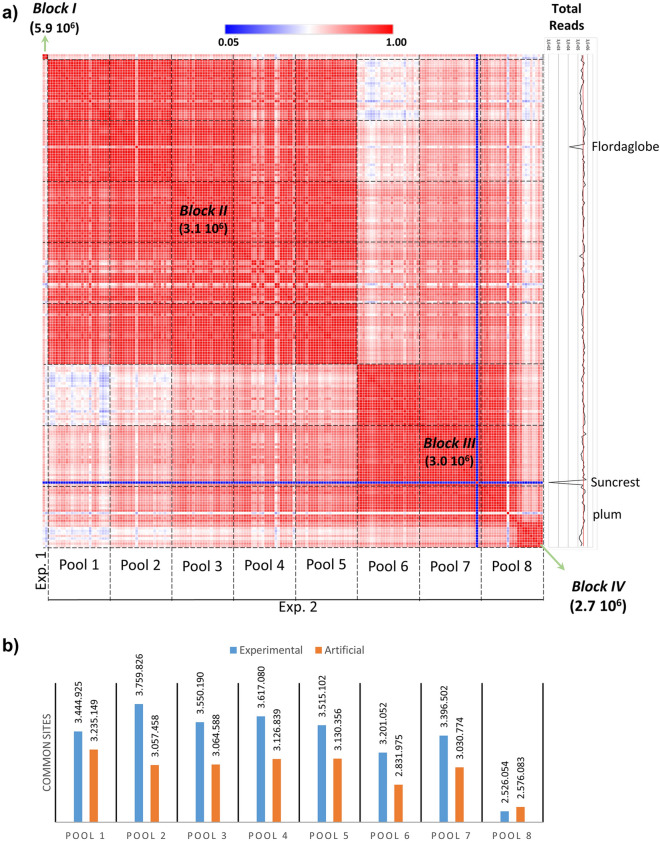
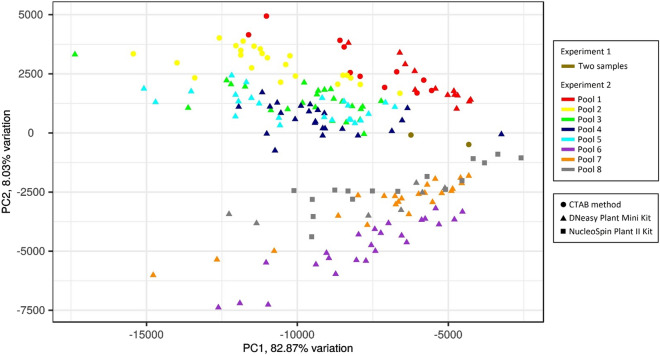
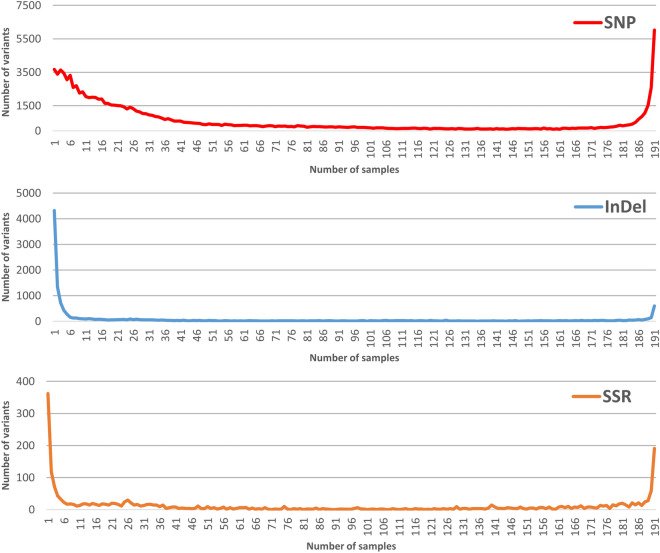
References
-
- FAO. http://faostat.fao.org. Accessed 27 Feb 2020 (2020).
-
- Gogorcena Y, Sánchez G, Moreno-Vázquez S, Pérez S, Ksouri N. Genomic-based breeding for climate-smart peach varieties. In: Kole C, editor. Genomic Designing of Climate-Smart Fruit Crops. Springer International Publishing; 2020. pp. 271–331.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
