

Gene replacement of α-globin with β-globin restores hemoglobin balance in β-thalassemia-derived hematopoietic stem and progenitor cells
- PMID: 33737751
- PMCID: PMC8265212
- DOI: 10.1038/s41591-021-01284-y
Gene replacement of α-globin with β-globin restores hemoglobin balance in β-thalassemia-derived hematopoietic stem and progenitor cells
Abstract
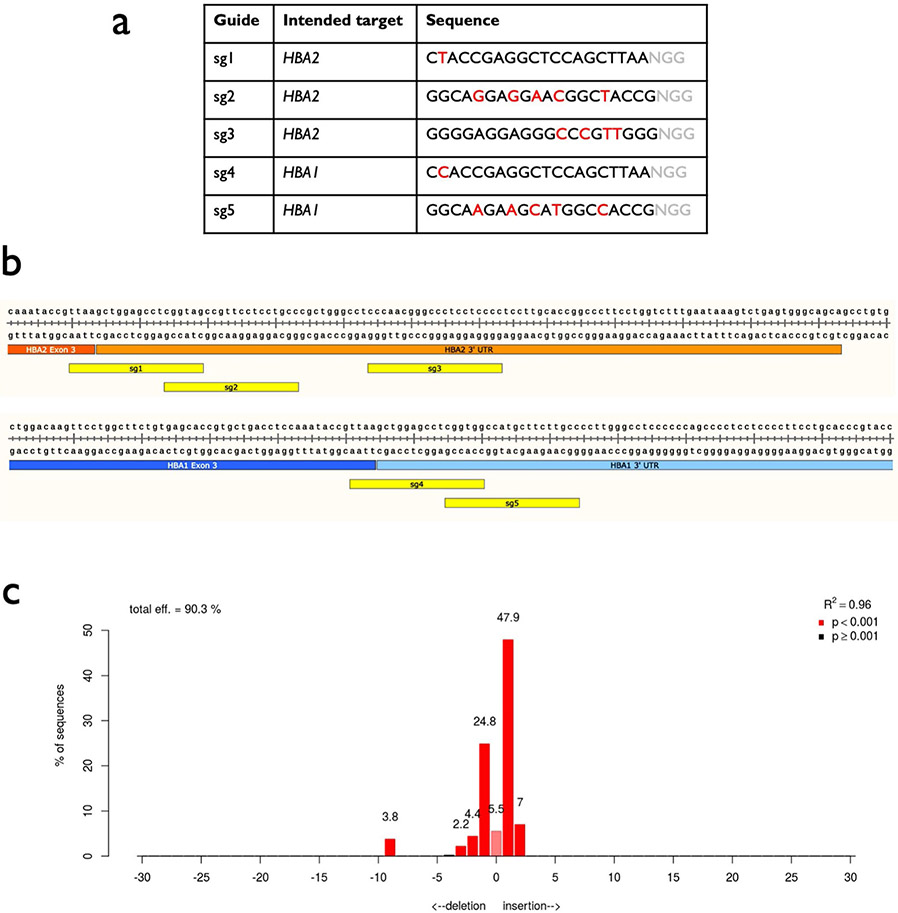
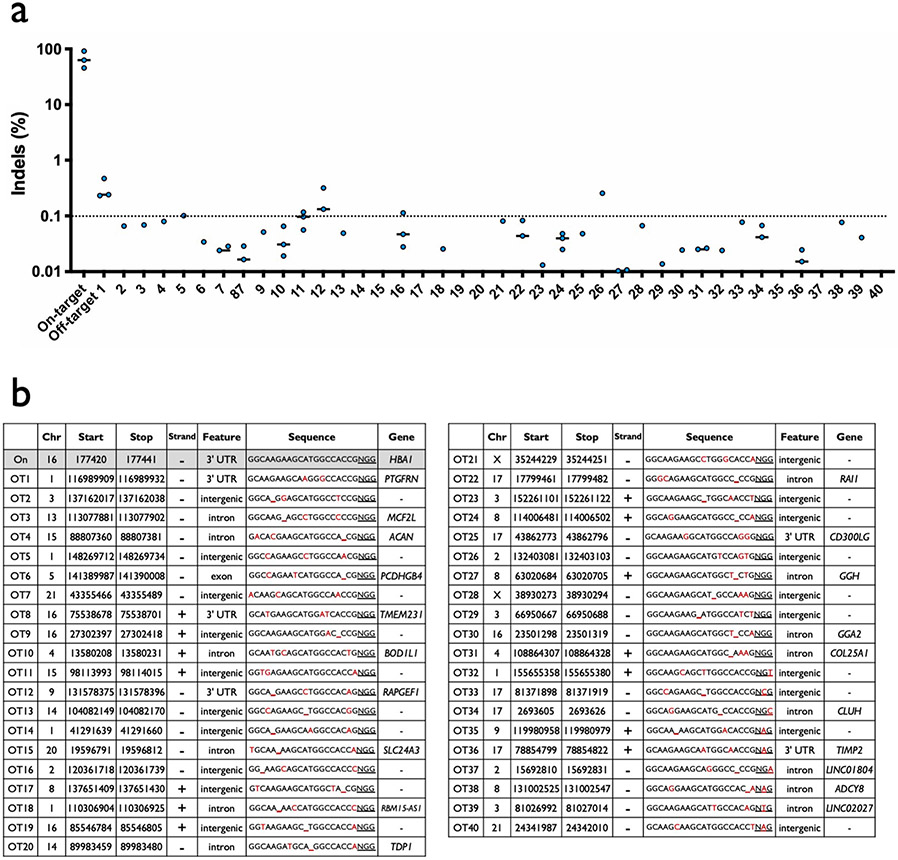
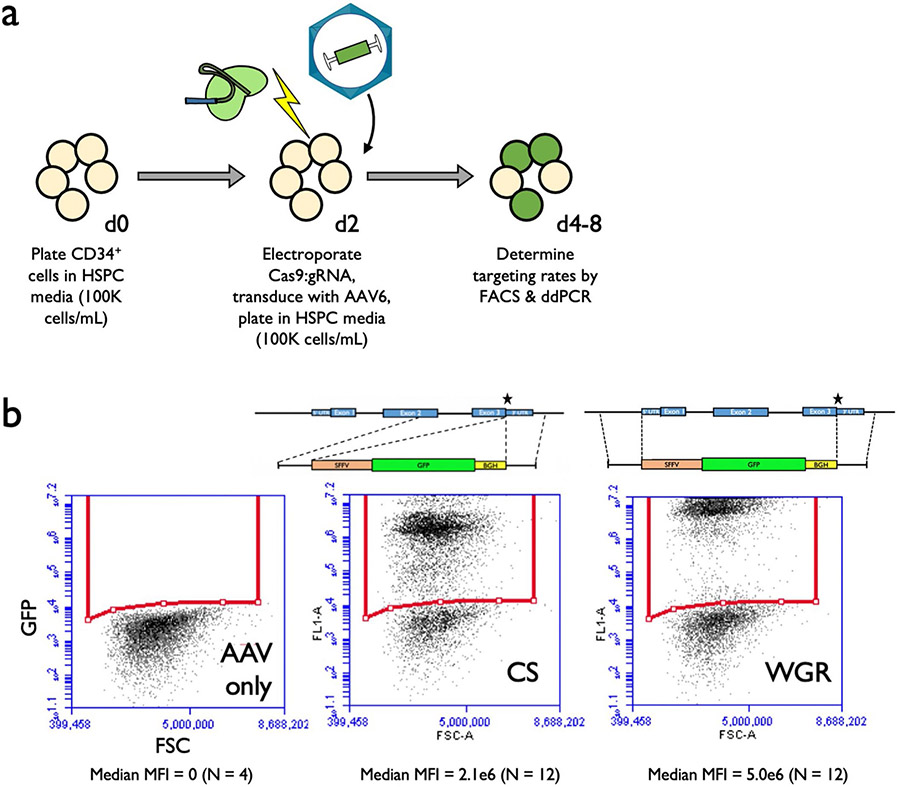
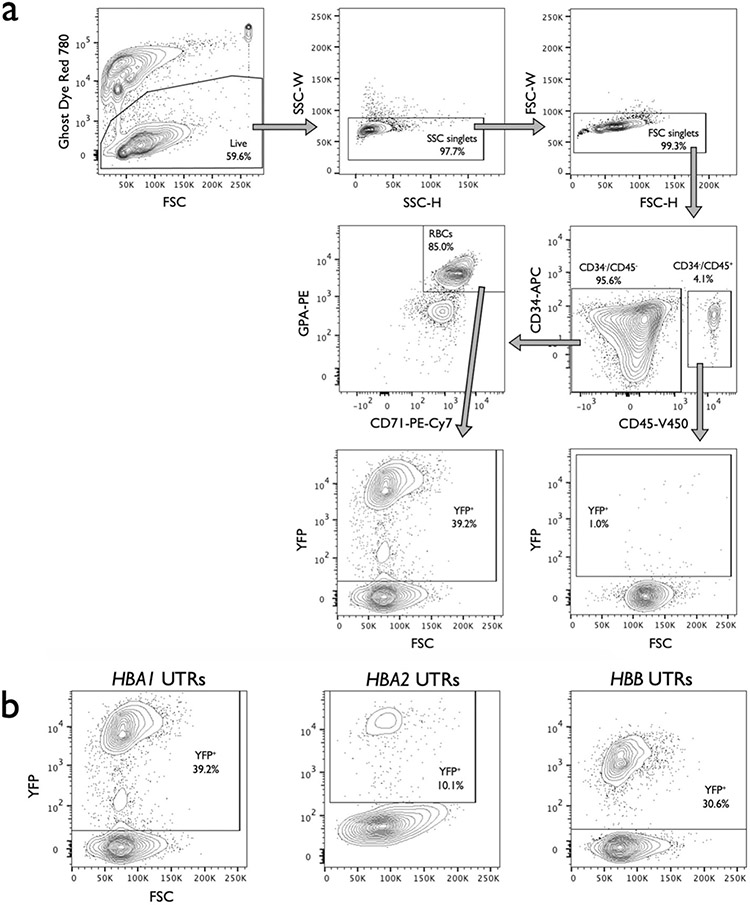
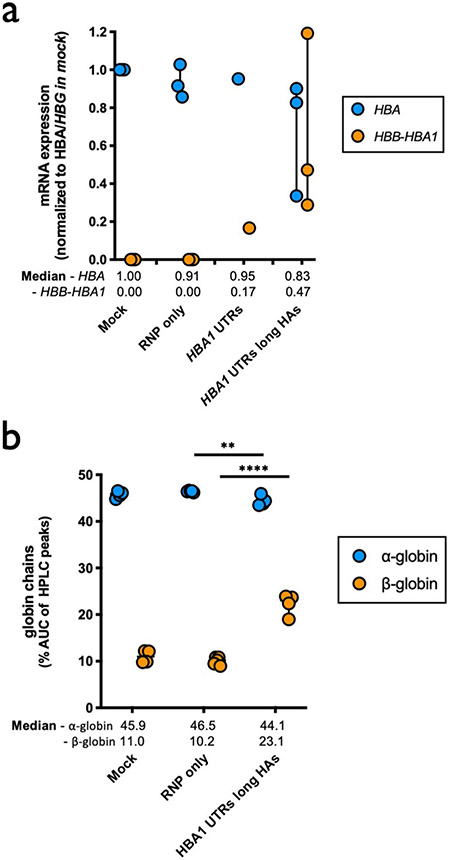
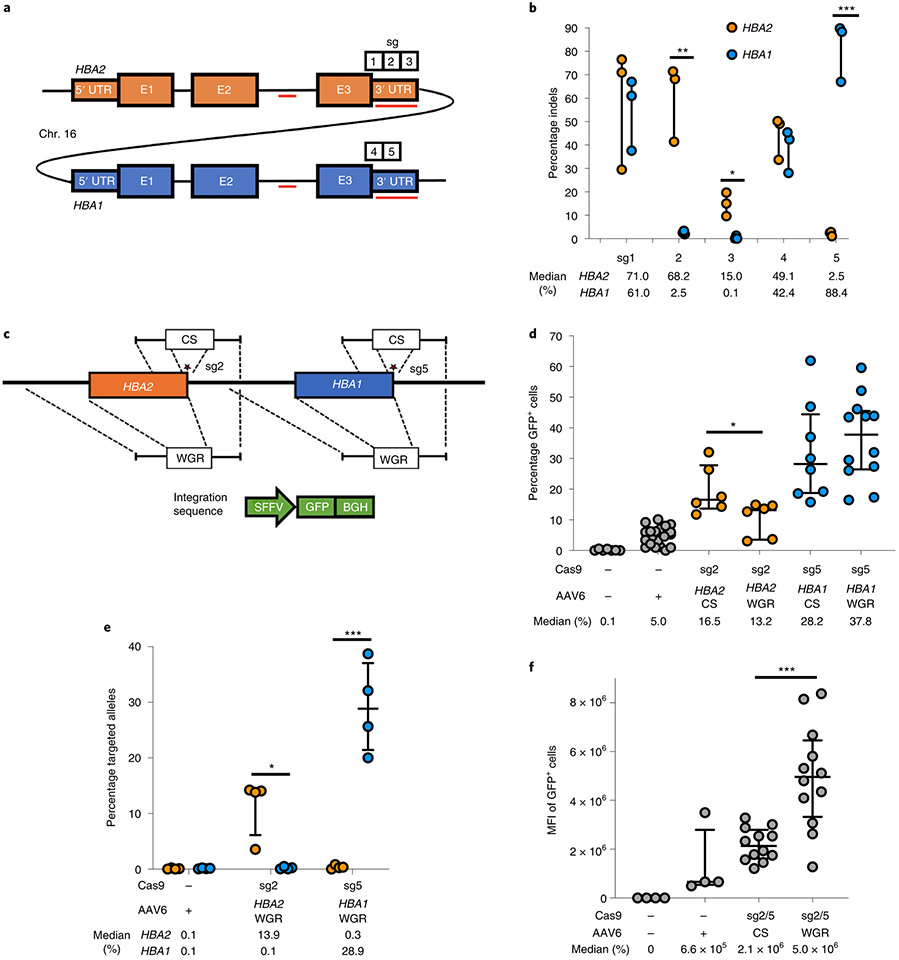
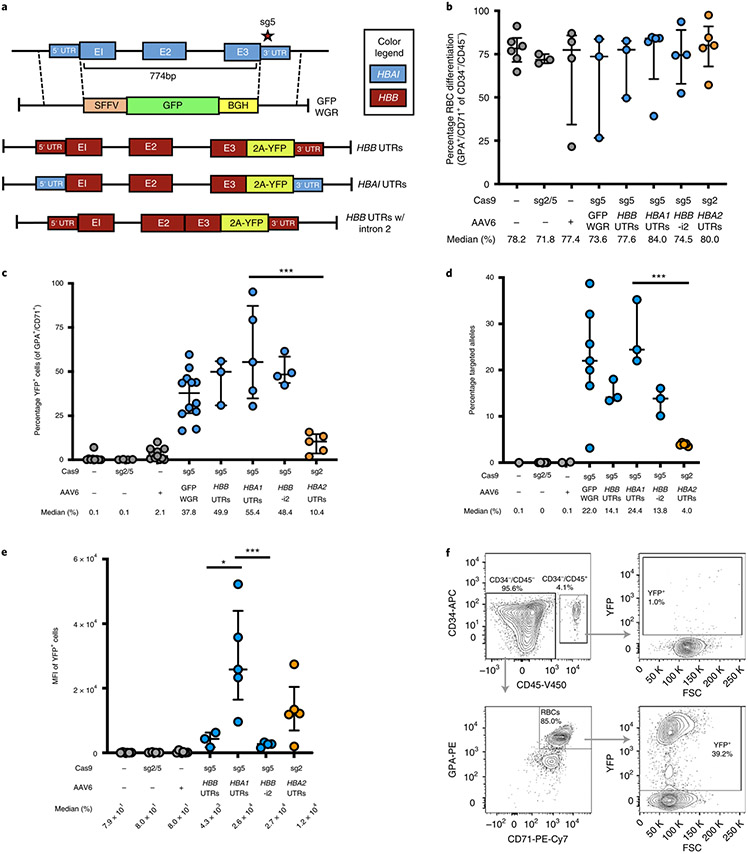
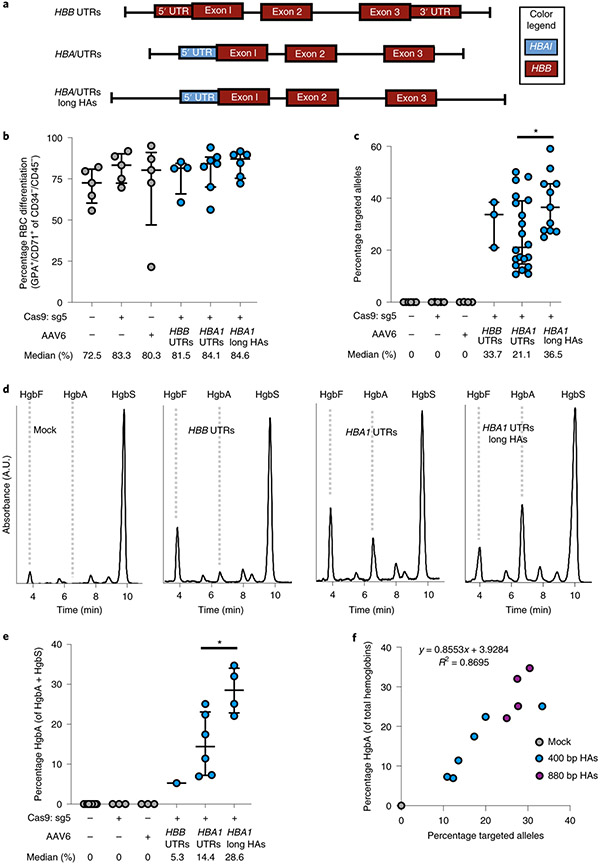
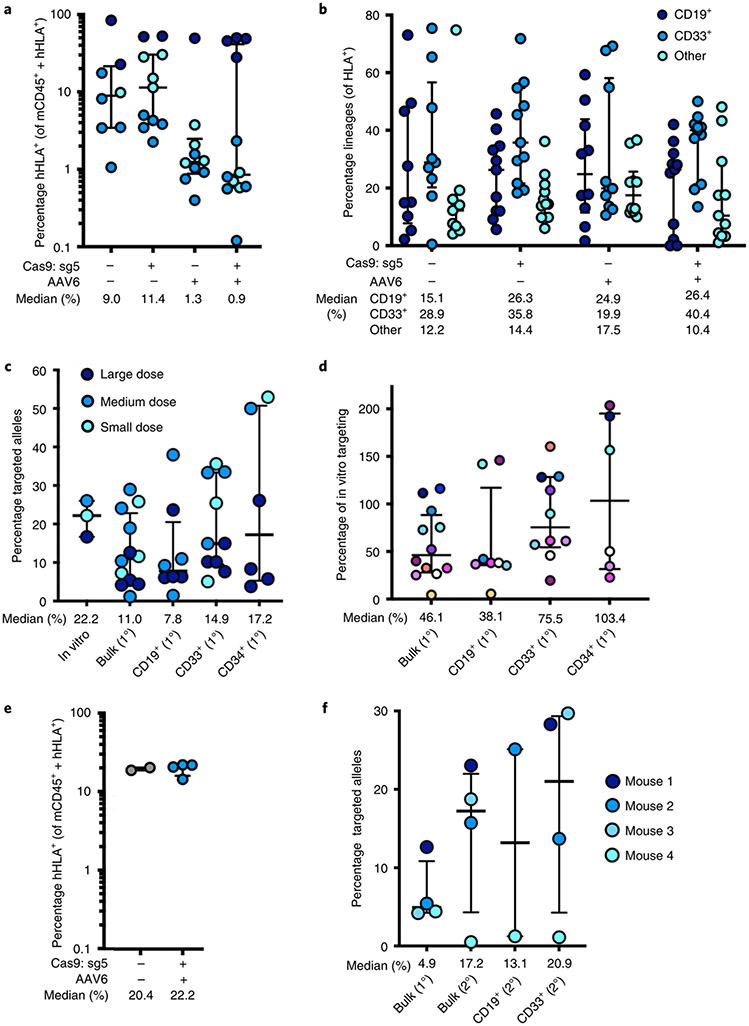
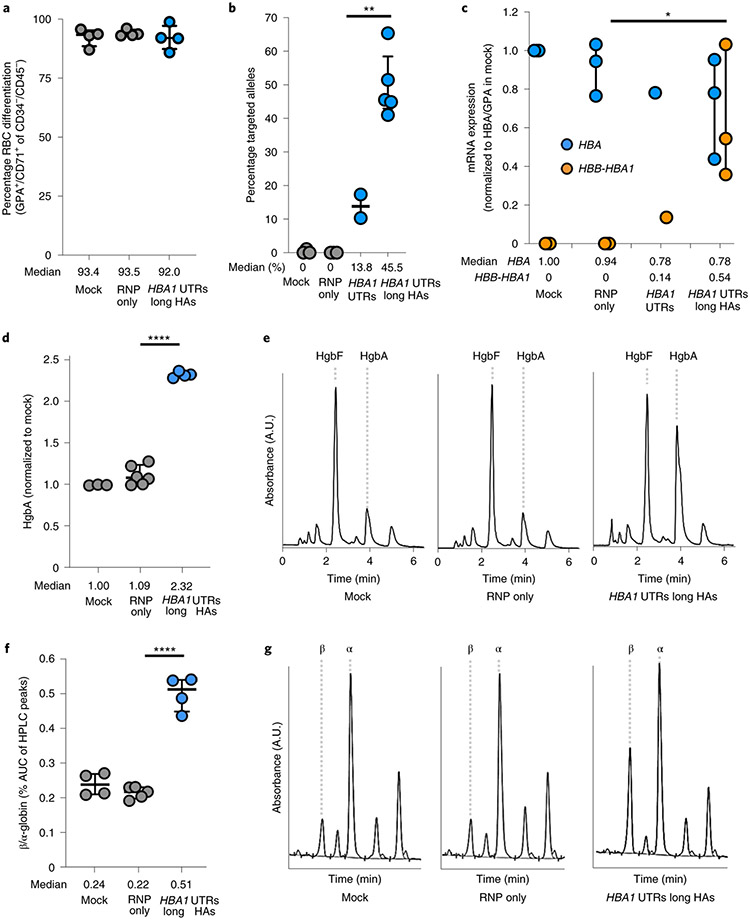
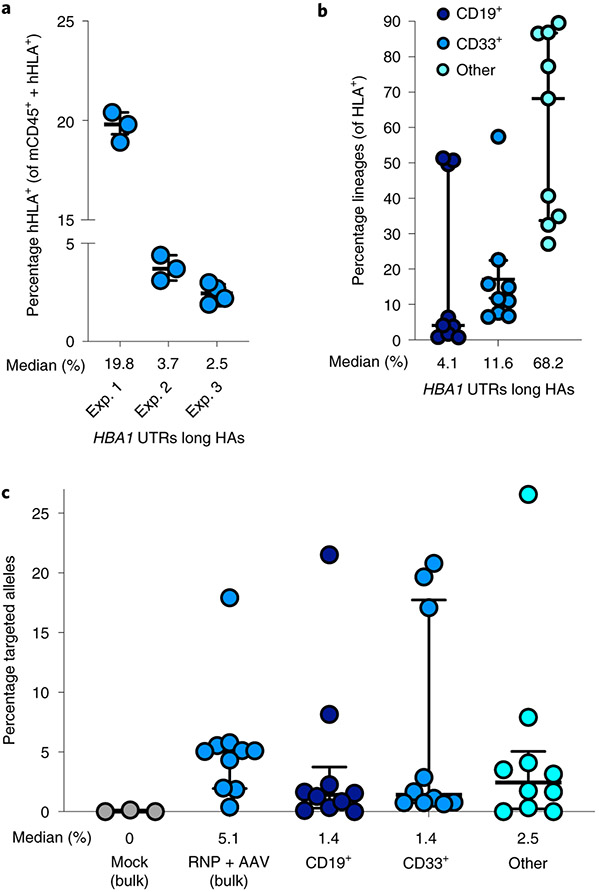
β-Thalassemia pathology is due not only to loss of β-globin (HBB), but also to erythrotoxic accumulation and aggregation of the β-globin-binding partner, α-globin (HBA1/2). Here we describe a Cas9/AAV6-mediated genome editing strategy that can replace the entire HBA1 gene with a full-length HBB transgene in β-thalassemia-derived hematopoietic stem and progenitor cells (HSPCs), which is sufficient to normalize β-globin:α-globin messenger RNA and protein ratios and restore functional adult hemoglobin tetramers in patient-derived red blood cells. Edited HSPCs were capable of long-term and bilineage hematopoietic reconstitution in mice, establishing proof of concept for replacement of HBA1 with HBB as a novel therapeutic strategy for curing β-thalassemia.
Figures
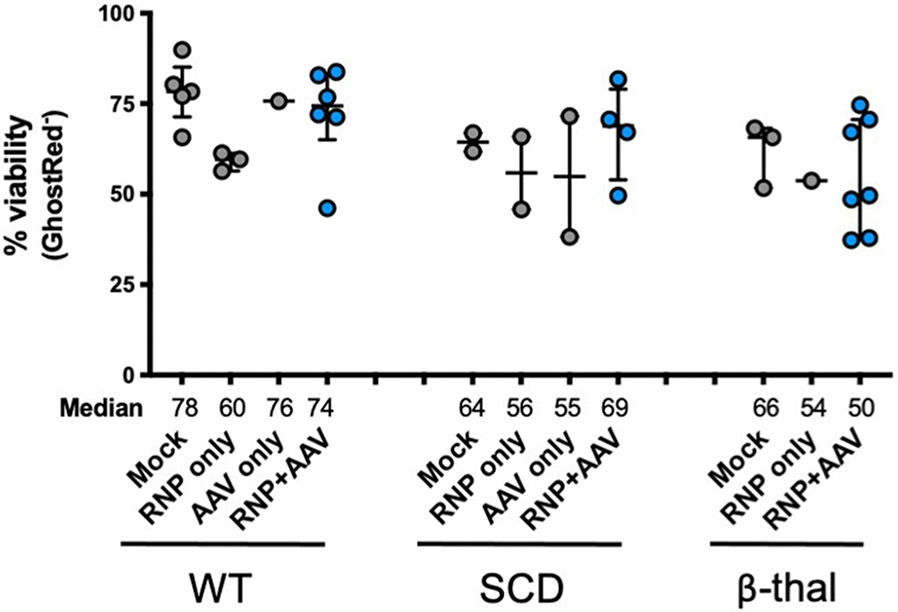
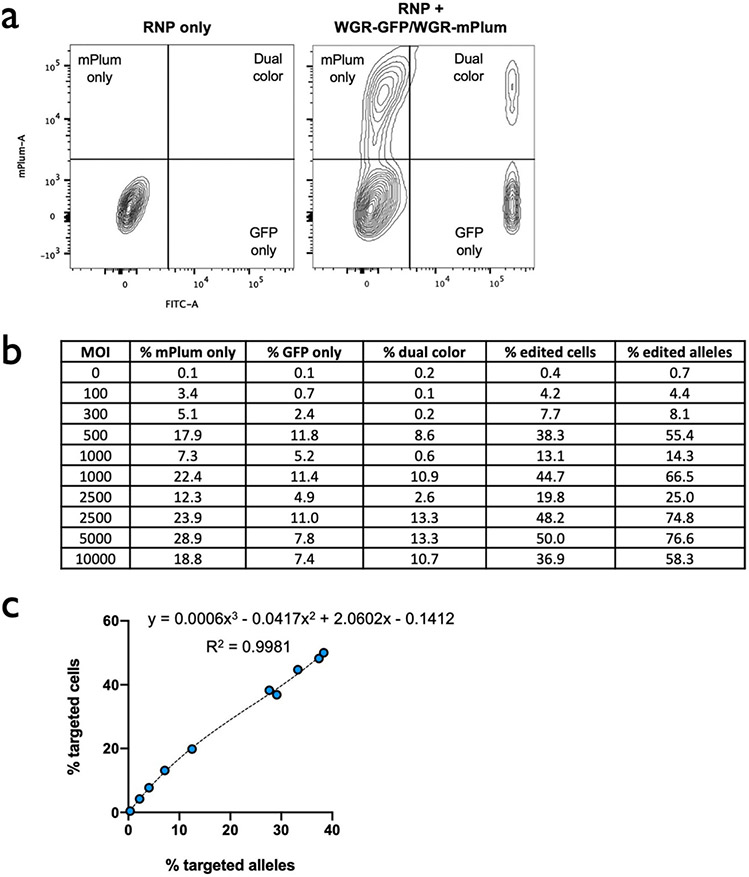
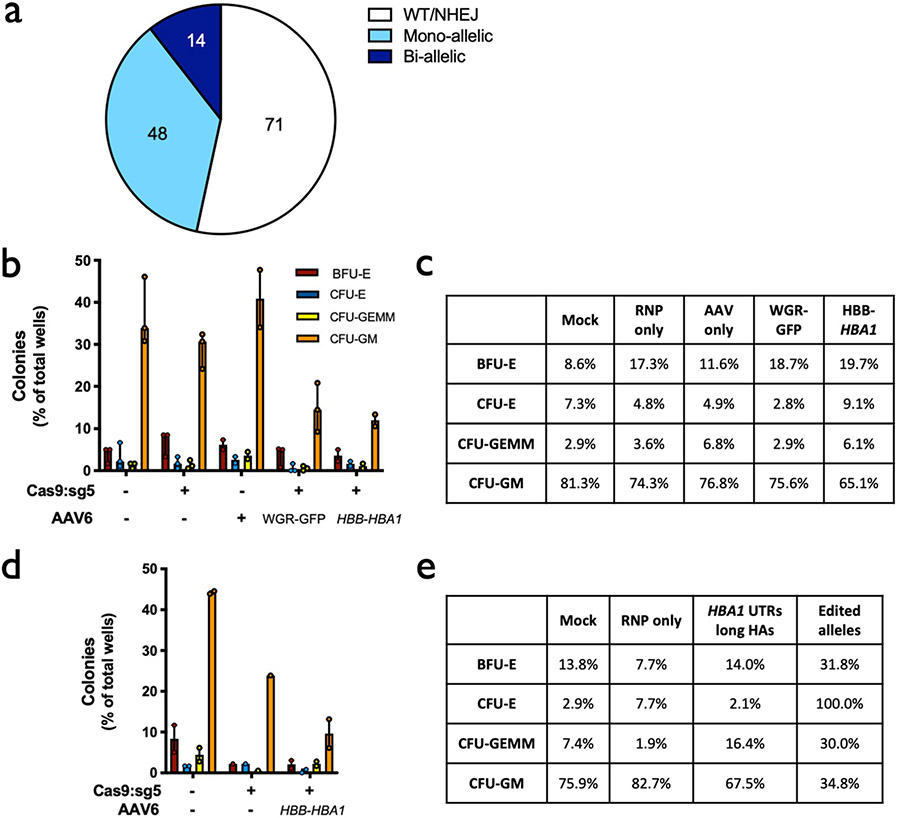
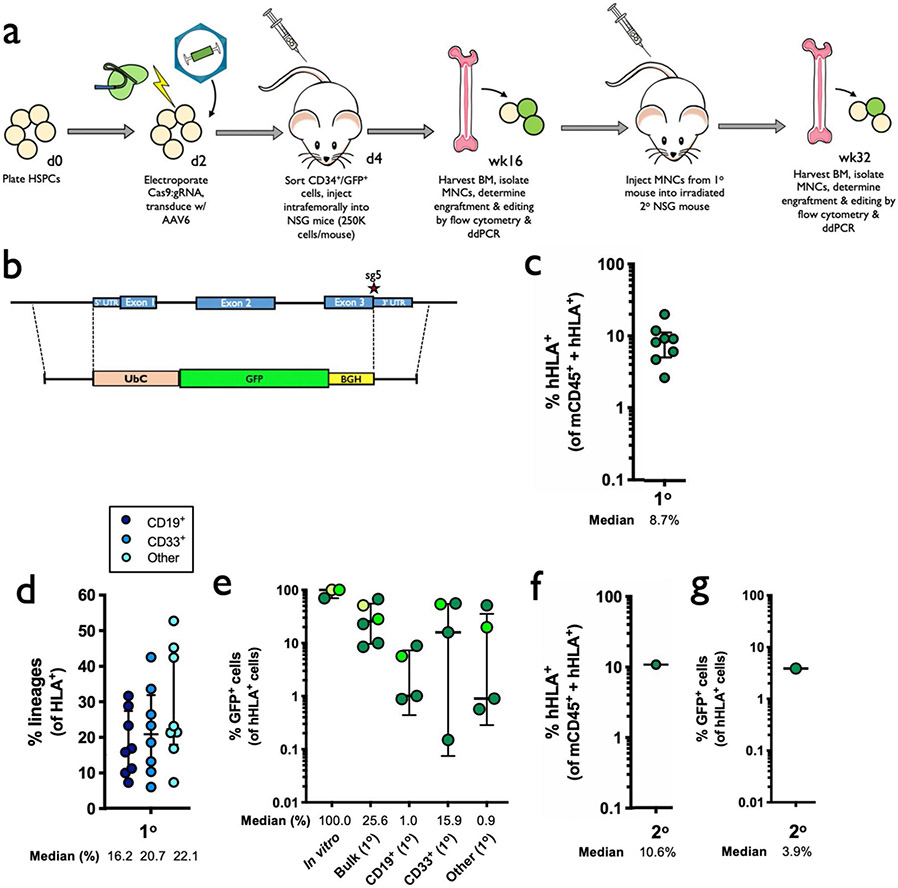
References
-
- Mentzer WC & Kan YW Prospects for research in hematologic disorders: sickle cell disease and thalassemia. JAMA 285, 640–642 (2001). - PubMed
-
- Ehlers KH, Giardina PJ, Lesser ML, Engle MA & Hilgartner MW Prolonged survival in patients with beta-thalassemia major treated with deferoxamine. J. Pediatr 118, 540–545 (1991). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
