

From Genotype to Phenotype: Expanding the Clinical Spectrum of CACNA1A Variants in the Era of Next Generation Sequencing
- PMID: 33737904
- PMCID: PMC7960780
- DOI: 10.3389/fneur.2021.639994
From Genotype to Phenotype: Expanding the Clinical Spectrum of CACNA1A Variants in the Era of Next Generation Sequencing
Abstract
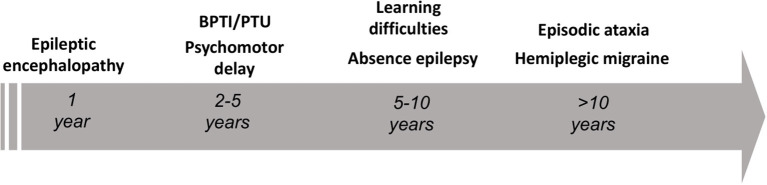
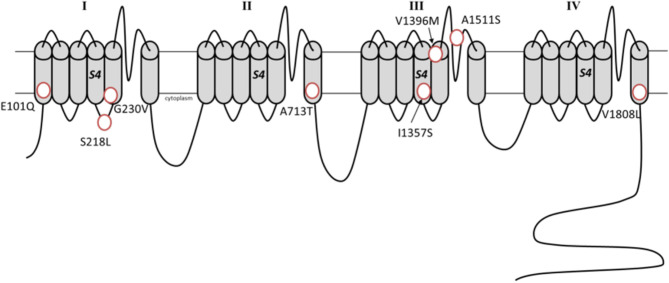
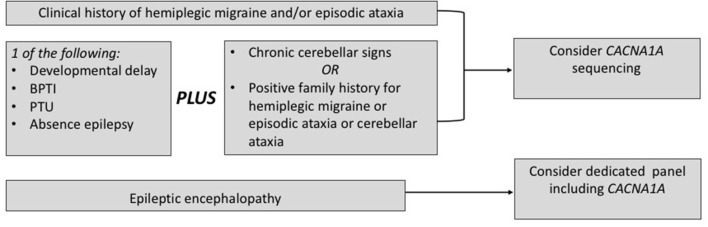
Ion channel dysfunction is a key pathological substrate of episodic neurological disorders. A classical gene associated to paroxysmal movement disorders is CACNA1A, which codes for the pore-forming subunit of the neuronal calcium channel P/Q. Non-polyglutamine CACNA1A variants underlie familial hemiplegic ataxia type 1 (FHM1) and episodic ataxia type 2 (EA2). Classical paroxysmal manifestations of FHM1 are migraine attacks preceded by motor aura consisting of hemiparesis, aphasia, and disturbances of consciousness until coma. Patients with EA2 suffer of recurrent episodes of vertigo, unbalance, diplopia, and vomiting. Beyond these typical presentations, several reports highlighted manifold clinical features associated with P/Q channelopathies, from chronic progressive cerebellar ataxia to epilepsy and psychiatric disturbances. These manifestations may often outlast the burden of classical episodic symptoms leading to pitfalls in the diagnostic work-up. Lately, the spreading of next generation sequencing techniques linked de novo CACNA1A variants to an even broader phenotypic spectrum including early developmental delay, autism spectrum disorders, epileptic encephalopathy, and early onset paroxysmal dystonia. The age-dependency represents a striking new aspect of these phenotypes und highlights a pivotal role for P/Q channels in the development of the central nervous system in a defined time window. While several reviews addressed the clinical presentation and treatment of FHM1 and EA2, an overview of the newly described age-dependent manifestations is lacking. In this Mini-Review we present a clinical update, delineate genotype-phenotype correlations as well as summarize evidence on the pathophysiological mechanisms underlying the expanded phenotype associated with CACNA1A variants.
Keywords: CACNA1A; calcium channels; de novo mutation; developmental delay; dystonia; epileptic encephalopathy; next generation sequencing; psychiatric manifestations.
Copyright © 2021 Indelicato and Boesch.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous