

Identification of Deep-Intronic Splice Mutations in a Large Cohort of Patients With Inherited Retinal Diseases
- PMID: 33737949
- PMCID: PMC7960924
- DOI: 10.3389/fgene.2021.647400
Identification of Deep-Intronic Splice Mutations in a Large Cohort of Patients With Inherited Retinal Diseases
Abstract
High throughput sequencing technologies have revolutionized the identification of mutations responsible for a diverse set of Mendelian disorders, including inherited retinal disorders (IRDs). However, the causal mutations remain elusive for a significant proportion of patients. This may be partially due to pathogenic mutations located in non-coding regions, which are largely missed by capture sequencing targeting the coding regions. The advent of whole-genome sequencing (WGS) allows us to systematically detect non-coding variations. However, the interpretation of these variations remains a significant bottleneck. In this study, we investigated the contribution of deep-intronic splice variants to IRDs. WGS was performed for a cohort of 571 IRD patients who lack a confident molecular diagnosis, and potential deep intronic variants that affect proper splicing were identified using SpliceAI. A total of six deleterious deep intronic variants were identified in eight patients. An in vitro minigene system was applied to further validate the effect of these variants on the splicing pattern of the associated genes. The prediction scores assigned to splice-site disruption positively correlated with the impact of mutations on splicing, as those with lower prediction scores demonstrated partial splicing. Through this study, we estimated the contribution of deep-intronic splice mutations to unassigned IRD patients and leveraged in silico and in vitro methods to establish a framework for prioritizing deep intronic variant candidates for mechanistic and functional analyses.
Keywords: deep-intronic mutations; inherited retinal dystrophies; minigenes; splicing; whole-genome sequencing.
Copyright © 2021 Qian, Wang, Wang, Igelman, Jones, Li, Wang, Goetz, Birch, Yang, Pennesi and Chen.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
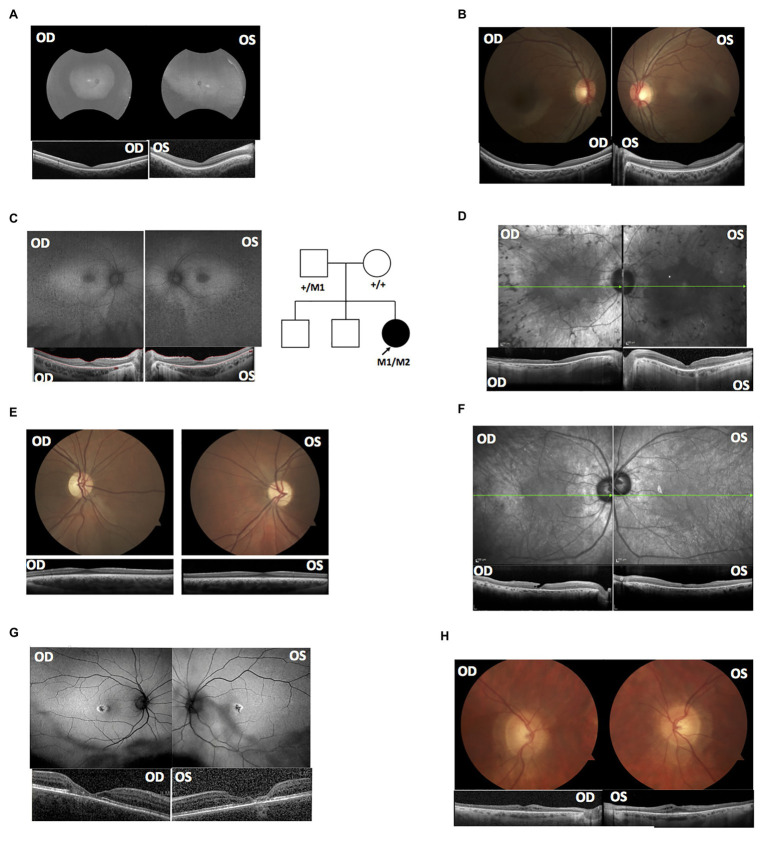

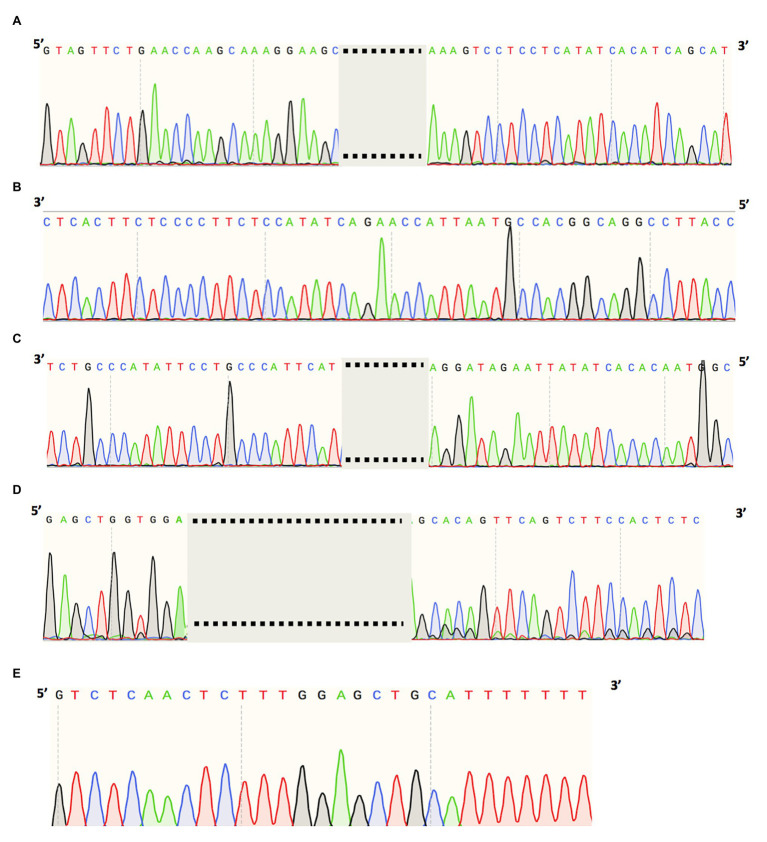
References
-
- Albert S., Garanto A., Sangermano R., Khan M., Bax N. M., Hoyng C. B., et al. (2018). Identification and rescue of splice defects caused by two neighboring deep-intronic ABCA4 mutations underlying Stargardt disease. Am. J. Hum. Genet. 102, 517–527. 10.1016/j.ajhg.2018.02.008, PMID: - DOI - PMC - PubMed
-
- Bauwens M., Garanto A., Sangermano R., Naessens S., Weisschuh N., De Zaeytijd J., et al. (2019). ABCA4-associated disease as a model for missing heritability in autosomal recessive disorders: novel noncoding splice, cis-regulatory, structural, and recurrent hypomorphic variants. Genet. Med. 21, 1761–1771. 10.1038/s41436-018-0420-y, PMID: - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources