

SARS-CoV-2 outbreak in a tri-national urban area is dominated by a B.1 lineage variant linked to a mass gathering event
- PMID: 33740028
- PMCID: PMC8011817
- DOI: 10.1371/journal.ppat.1009374
SARS-CoV-2 outbreak in a tri-national urban area is dominated by a B.1 lineage variant linked to a mass gathering event
Abstract
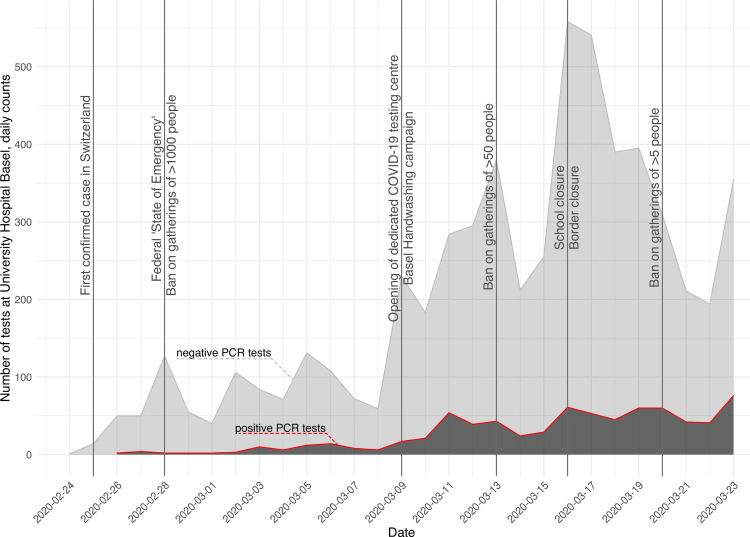
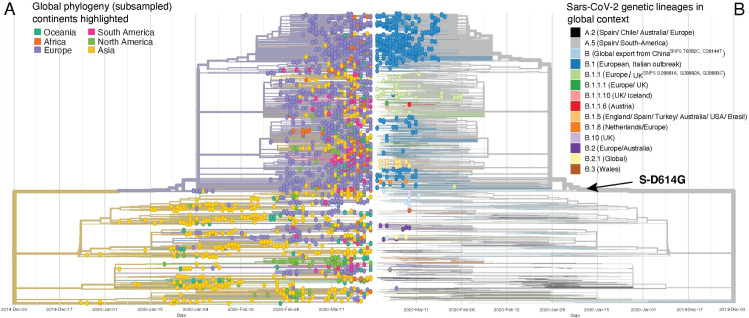
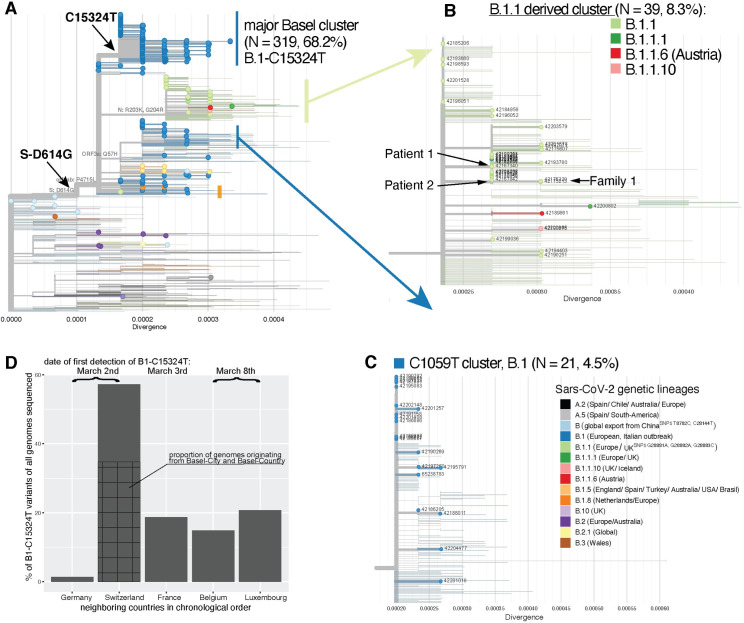
The first case of SARS-CoV-2 in Basel, Switzerland was detected on February 26th 2020. We present a phylogenetic study to explore viral introduction and evolution during the exponential early phase of the local COVID-19 outbreak from February 26th until March 23rd. We sequenced SARS-CoV-2 naso-oropharyngeal swabs from 746 positive tests that were performed at the University Hospital Basel during the study period. We successfully generated 468 high quality genomes from unique patients and called variants with our COVID-19 Pipeline (COVGAP), and analysed viral genetic diversity using PANGOLIN taxonomic lineages. To identify introduction and dissemination events we incorporated global SARS-CoV-2 genomes and inferred a time-calibrated phylogeny. Epidemiological data from patient questionnaires was used to facilitate the interpretation of phylogenetic observations. The early outbreak in Basel was dominated by lineage B.1 (83·6%), detected first on March 2nd, although the first sample identified belonged to B.1.1. Within B.1, 68·2% of our samples fall within a clade defined by the SNP C15324T ('Basel cluster'), including 157 identical sequences at the root of the 'Basel cluster', some of which we can specifically trace to regional spreading events. We infer the origin of B.1-C15324T to mid-February in our tri-national region. The other genomes map broadly over the global phylogenetic tree, showing several introduction events from and/or dissemination to other regions of the world via travellers. Family transmissions can also be traced in our data. A single lineage variant dominated the outbreak in the Basel area while other lineages, such as the first (B.1.1), did not propagate. A mass gathering event was the predominant initial source of cases, with travel returners and family transmissions to a lesser extent. We highlight the importance of adding specific questions to epidemiological questionnaires, to obtain data on attendance of large gatherings and their locations, as well as travel history, to effectively identify routes of transmissions in up-coming outbreaks. This phylogenetic analysis in concert with epidemiological and contact tracing data, allows connection and interpretation of events, and can inform public health interventions. Trial Registration: ClinicalTrials.gov NCT04351503.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

References
MeSH terms
Associated data
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
