

Epigenetic mechanisms in breast cancer therapy and resistance
- PMID: 33741974
- PMCID: PMC7979820
- DOI: 10.1038/s41467-021-22024-3
Epigenetic mechanisms in breast cancer therapy and resistance
Abstract
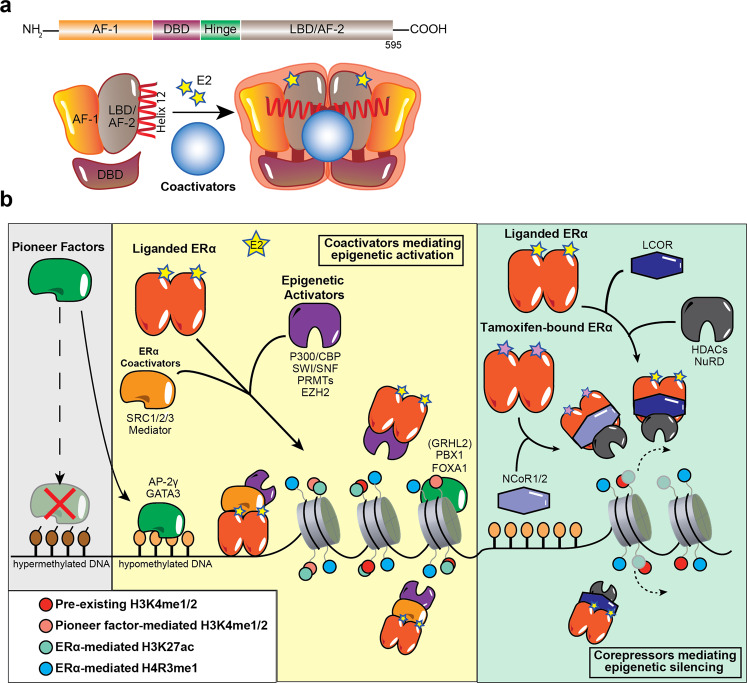
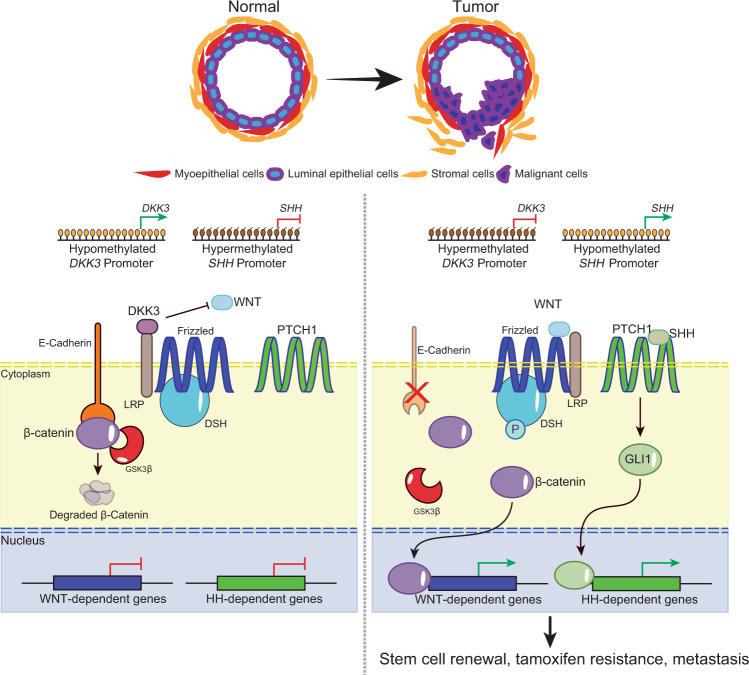
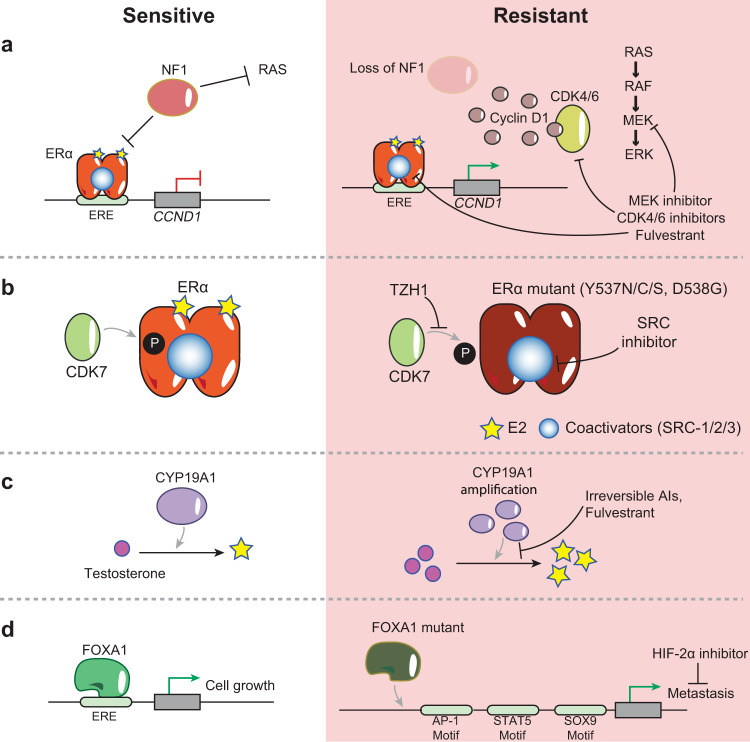
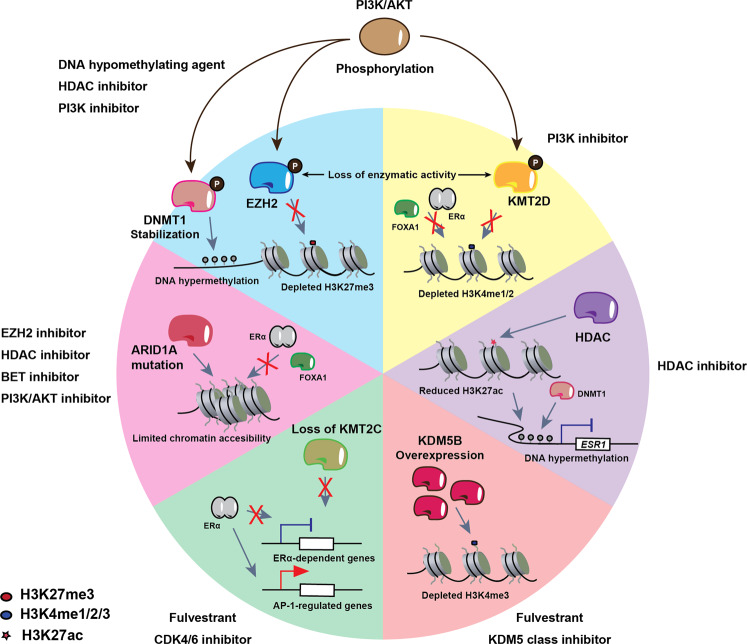
The majority of breast cancers express the estrogen receptor (ERα) and agents targeting this pathway represent the main treatment modality. Endocrine therapy has proven successful in the treatment of hormone-responsive breast cancer since its early adoption in the 1940s as an ablative therapy. Unfortunately, therapeutic resistance arises, leading to disease recurrence and relapse. Recent studies increased our understanding in how changes to the chromatin landscape and deregulation of epigenetic factors orchestrate the resistant phenotype. Here, we will discuss how the epigenome is an integral determinant in hormone therapy response and why epigenetic factors are promising targets for overcoming clinical resistance.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Deblois G, et al. Epigenetic switch–induced viral mimicry evasion in chemotherapy-resistant breast cancer. Cancer Discov. 2020;10:1312–1329. doi: 10.1158/2159-8290.CD-19-1493. - DOI - PubMed
-
- American Cancer Society. Breast cancer facts and figures 2019–2020, (American Cancer Society, 2019).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
