

Inflation of tumor mutation burden by tumor-only sequencing in under-represented groups
- PMID: 33742076
- PMCID: PMC7979755
- DOI: 10.1038/s41698-021-00164-5
Inflation of tumor mutation burden by tumor-only sequencing in under-represented groups
Abstract
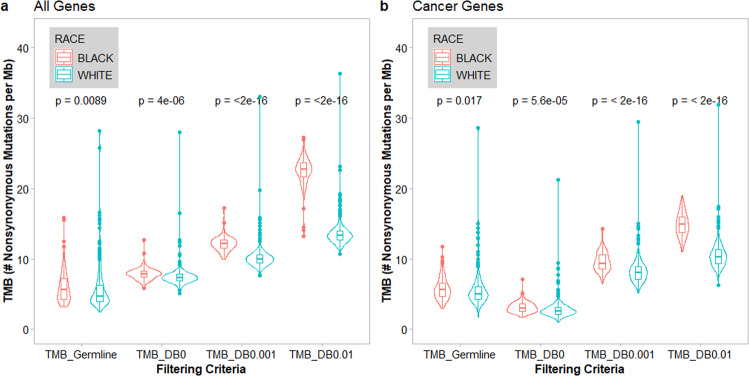
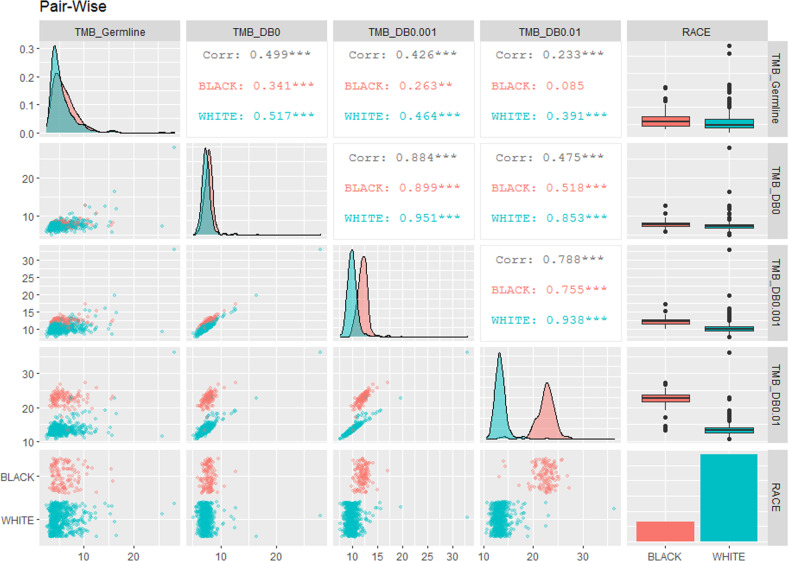
With the recent FDA approval of tumor mutational burden-high (TMB-H) status as a biomarker for treatment with a PD-1 inhibitor regardless of tumor type, accurate assessment of patient-specific TMB is more critical now more than ever. Using paired tumor and germline exome sequencing data from 701 patients newly diagnosed with multiple myeloma, including 575 self-reported White patients and 126 self-reported Black patients, we observed that compared to the gold standard of filtering germline variants with patient-paired germline sequencing data, TMB estimates were significantly higher in both Black and White patients when using public databases for filtering non-somatic mutations; however, TMB was more significantly inflated in Black patients compared to White patients. TMB as a biomarker for patient selection to receive immune checkpoint inhibitors (ICIs) therapy without patient-paired germline sequencing may introduce racial bias due to the under-representation of minority groups in public databases.
Conflict of interest statement
Dr. Mansfield reports research support from Novartis and Verily; remuneration to his institution for participation on advisory boards for AbbVie, Astra Zeneca, BMS, and Genentech; travel support from Roche, and is a non-remunerated director of the Mesothelioma Applied Research Foundation. Dr. Parikh reports serving on advisory boards for AstraZeneca and Blueprint Medicines. Dr. Board reports grant funding to institution from Senhwa Pharmaceuticals, Adaptimmune, Agios Pharmaceuticals, Halozyme Pharmaceuticals, Celgene Pharmaceuticals, EMD Merck Serono, Toray, Dicerna, Taiho Pharmaceuticals, Sun Biopharma, Isis Pharmaceuticals, Redhill Pharmaceuticals, Boston Biomed, Basilea, Incyte Pharmaceuticals, Mirna Pharmaceuticals, Medimmune, Bioline, Sillajen, ARIAD Pharmaceuticals, PUMA Pharmaceuticals, Novartis, QED Pharmaceuticals, Pieris Pharmaceuticals; consulting fees to self from ADC Therapeutics, Exelixis Pharmaceuticals, Inspyr Therapeutics, G1 Therapeutics, Immunovative Therapies, OncBioMune Pharmaceuticals, Western Oncolytics, Lynx Group, Genentech, Merck, Huya; travel support to self from AstraZeneca. All other authors have no relevant competing interests to declare.
Figures
References
-
- Marabelle A, et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020;21:1353–1365. doi: 10.1016/S1470-2045(20)30445-9. - DOI - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
