

Targeting chemokines for acute lymphoblastic leukemia therapy
- PMID: 33743810
- PMCID: PMC7981899
- DOI: 10.1186/s13045-021-01060-y
Targeting chemokines for acute lymphoblastic leukemia therapy
Abstract
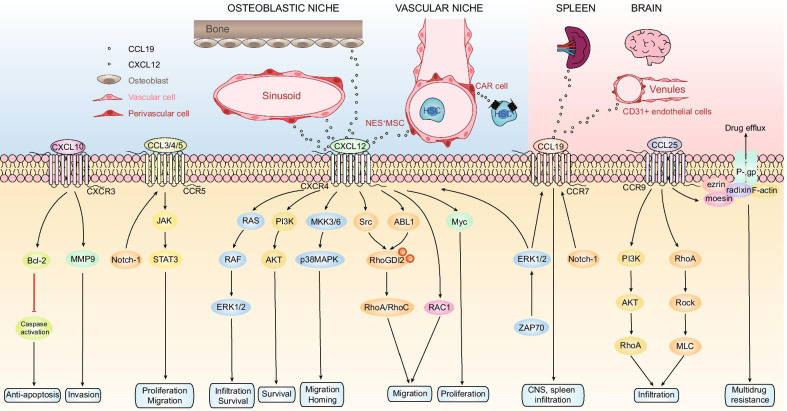
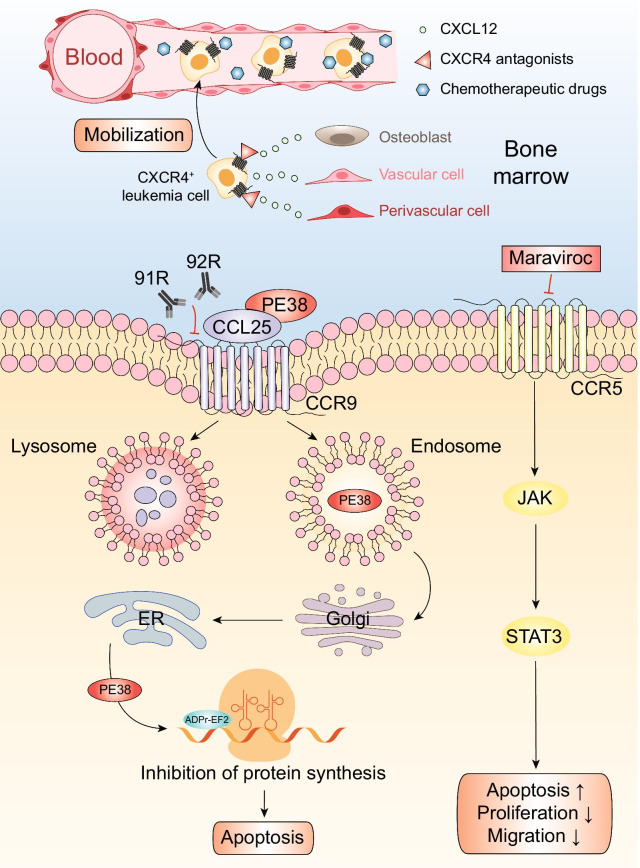
Acute lymphoblastic leukemia (ALL) is a hematological malignancy characterized by the malignant clonal expansion of lymphoid hematopoietic precursors. It is regulated by various signaling molecules such as cytokines and adhesion molecules in its microenvironment. Chemokines are chemotactic cytokines that regulate migration, positioning and interactions of cells. Many chemokine axes such as CXCL12/CXCR4 and CCL25/CCR9 have been proved to play important roles in leukemia microenvironment and further affect ALL outcomes. In this review, we summarize the chemokines that are involved in ALL progression and elaborate on their roles and mechanisms in leukemia cell proliferation, infiltration, drug resistance and disease relapse. We also discuss the potential of targeting chemokine axes for ALL treatments, since many related inhibitors have shown promising efficacy in preclinical trials, and some of them have entered clinical trials.
Keywords: Acute lymphoblastic leukemia; Chemokine; Microenvironment; Therapeutic targets.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Chiarini F, Lonetti A, Evangelisti C, Buontempo F, Orsini E, Evangelisti C, Cappellini A, Neri LM, McCubrey JA, Martelli AM. Advances in understanding the acute lymphoblastic leukemia bone marrow microenvironment: From biology to therapeutic targeting. Biochem Biophys Acta. 2016;1863(3):449–463. doi: 10.1016/j.bbamcr.2015.08.015. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
