

Sequential Administration of XPO1 and ATR Inhibitors Enhances Therapeutic Response in TP53-mutated Colorectal Cancer
- PMID: 33745946
- PMCID: PMC8238881
- DOI: 10.1053/j.gastro.2021.03.022
Sequential Administration of XPO1 and ATR Inhibitors Enhances Therapeutic Response in TP53-mutated Colorectal Cancer
Abstract
Background & aims: Understanding the mechanisms by which tumors adapt to therapy is critical for developing effective combination therapeutic approaches to improve clinical outcomes for patients with cancer.
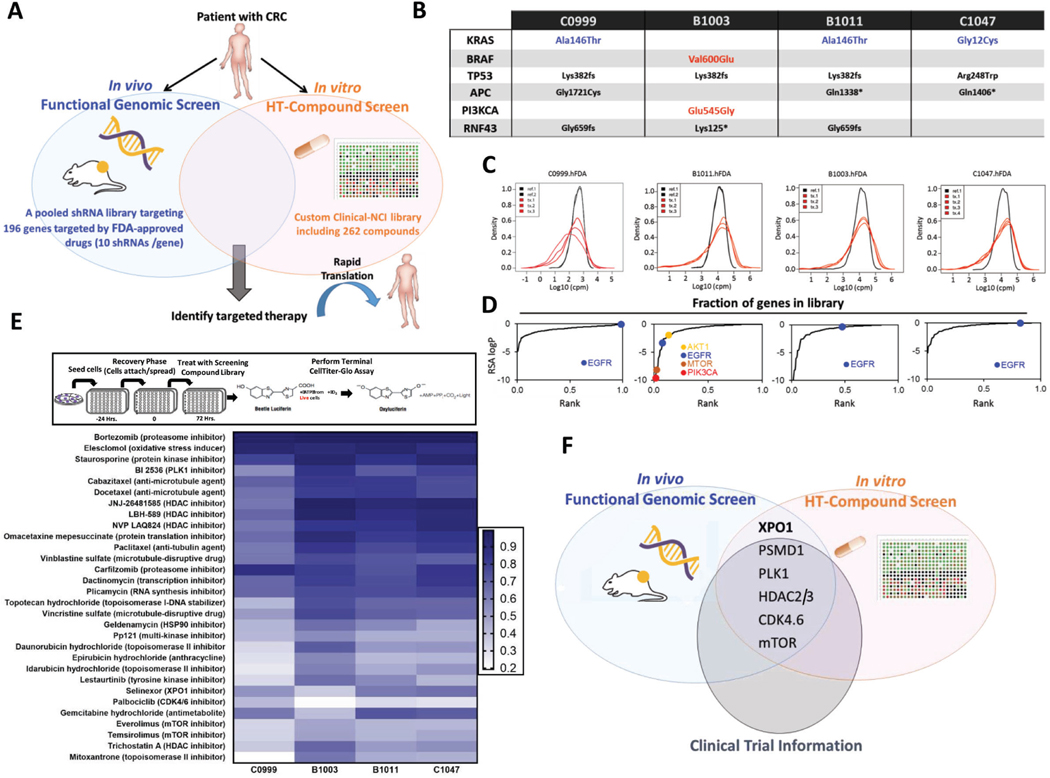
Methods: To identify promising and clinically actionable targets for managing colorectal cancer (CRC), we conducted a patient-centered functional genomics platform that includes approximately 200 genes and paired this with a high-throughput drug screen that includes 262 compounds in four patient-derived xenografts (PDXs) from patients with CRC.
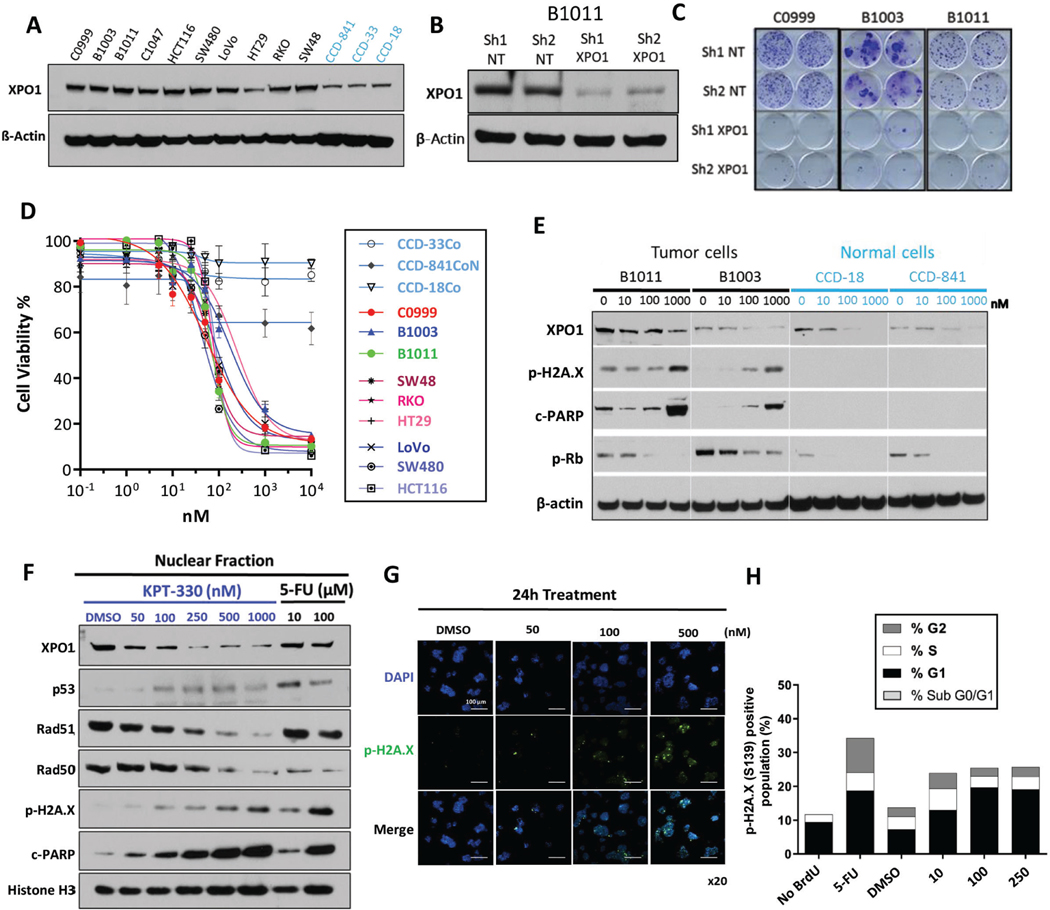
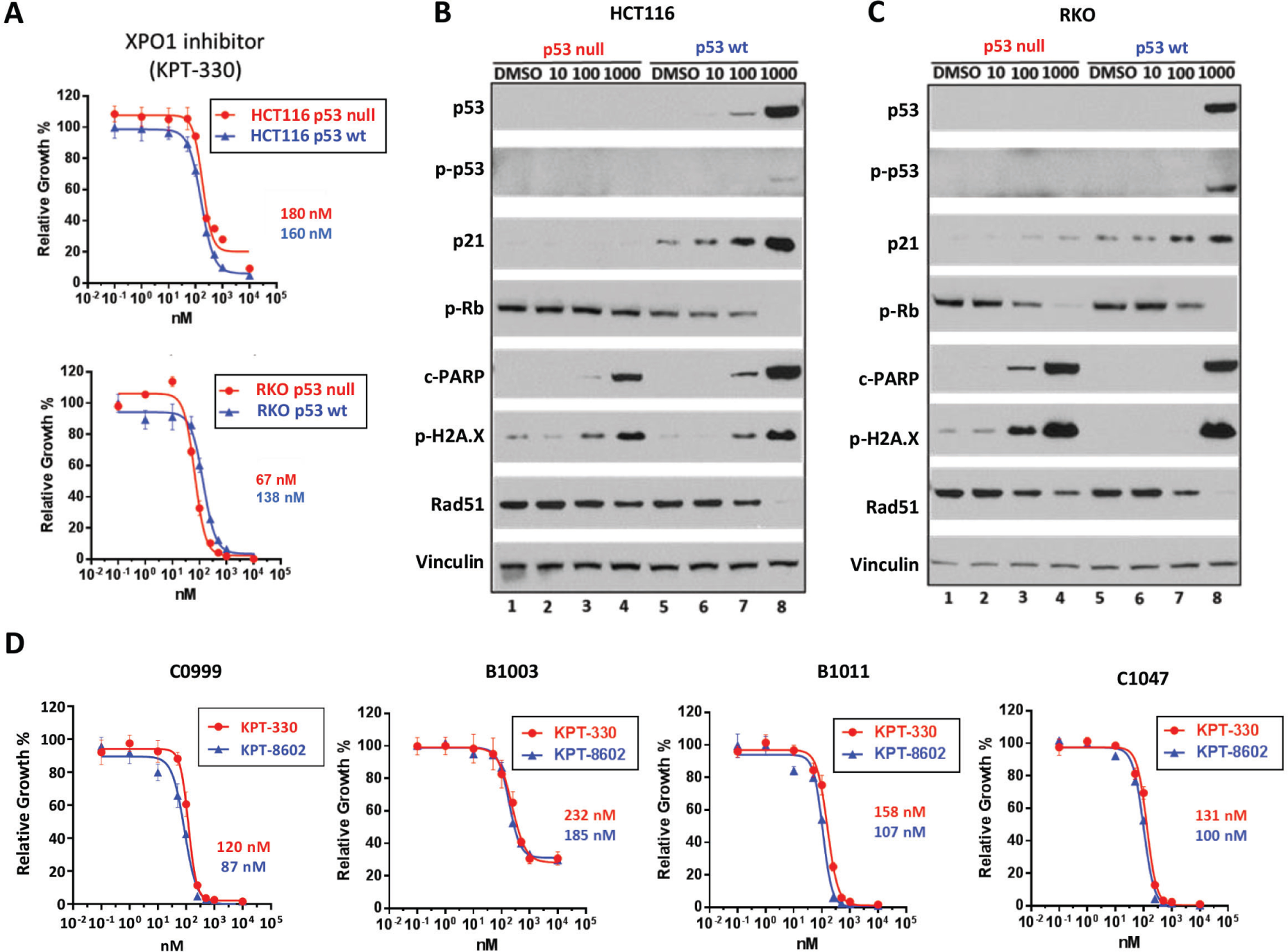
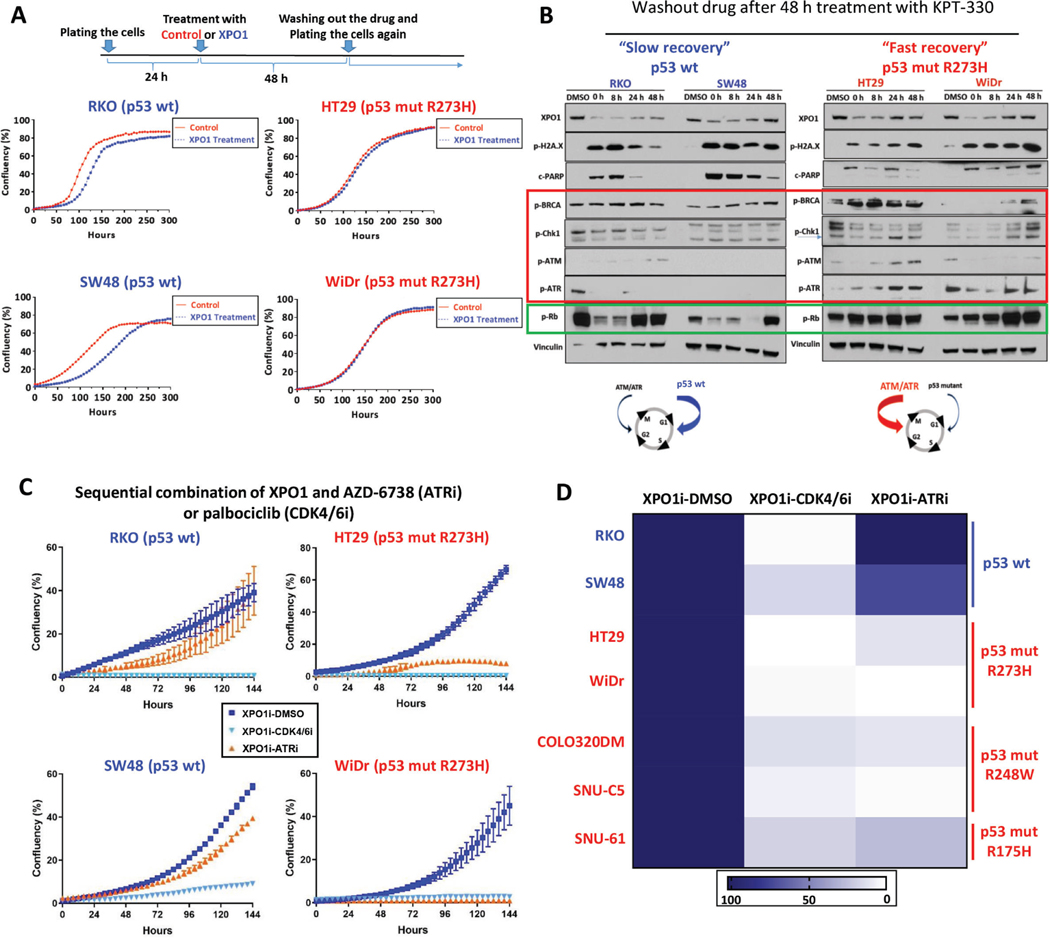
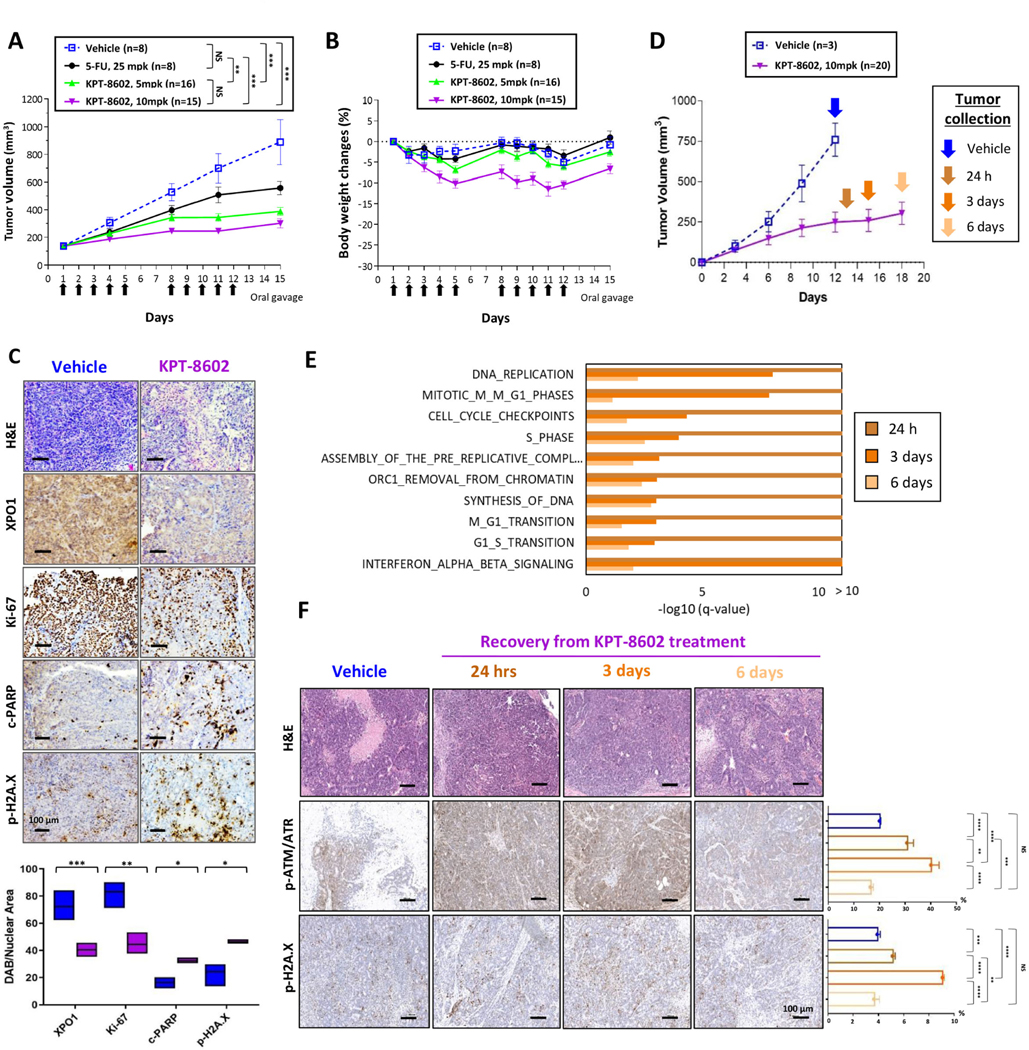
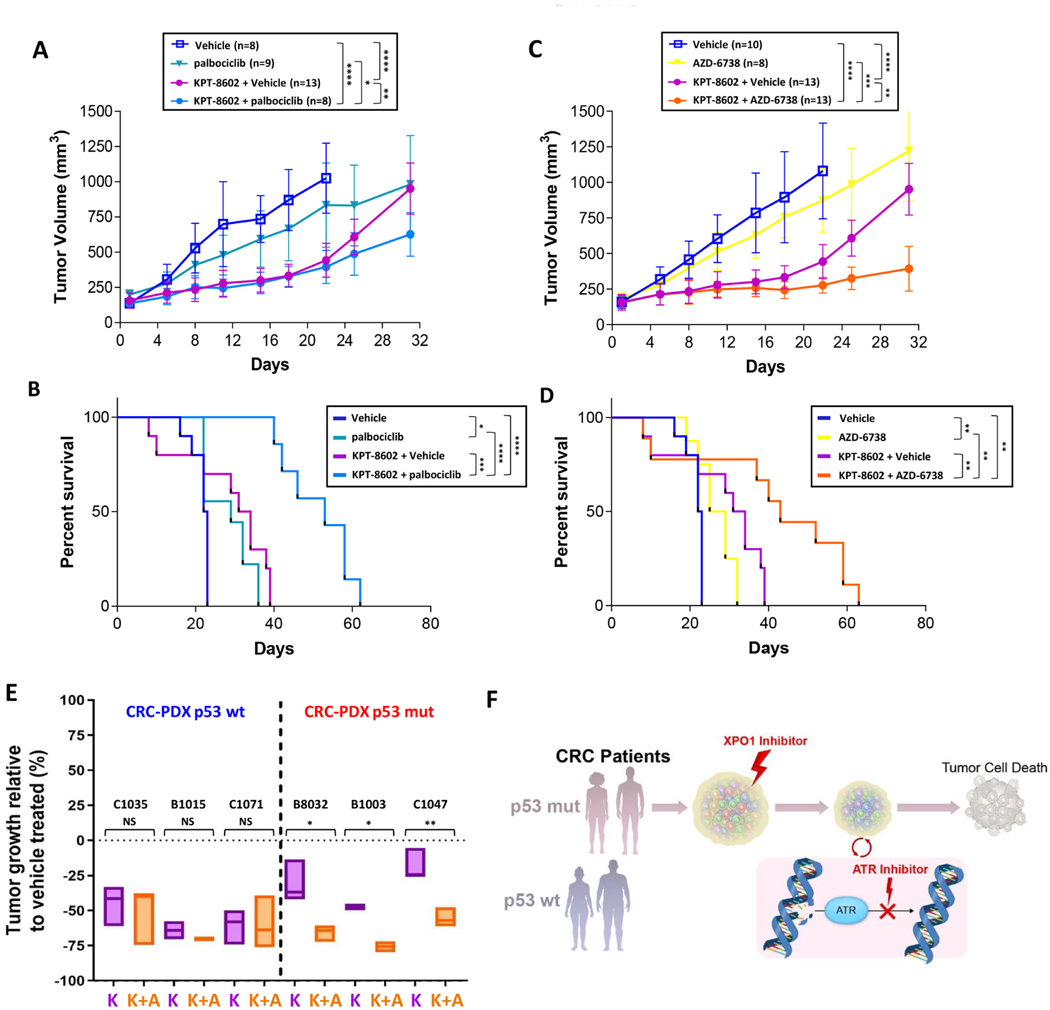
Results: Both screening methods identified exportin 1 (XPO1) inhibitors as drivers of DNA damage-induced lethality in CRC. Molecular characterization of the cellular response to XPO1 inhibition uncovered an adaptive mechanism that limited the duration of response in TP53-mutated, but not in TP53-wild-type CRC models. Comprehensive proteomic and transcriptomic characterization revealed that the ATM/ATR-CHK1/2 axes were selectively engaged in TP53-mutant CRC cells upon XPO1 inhibitor treatment and that this response was required for adapting to therapy and escaping cell death. Administration of KPT-8602, an XPO1 inhibitor, followed by AZD-6738, an ATR inhibitor, resulted in dramatic antitumor effects and prolonged survival in TP53-mutant models of CRC.
Conclusions: Our findings anticipate tremendous therapeutic benefit and support the further evaluation of XPO1 inhibitors, especially in combination with DNA damage checkpoint inhibitors, to elicit an enduring clinical response in patients with CRC harboring TP53 mutations.
Keywords: CRC; Combination Therapy; Genomic Biomarker; PDX.
Copyright © 2021 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Figures
Comment in
-
Trapping Colorectal Cancer Into a Dead-end.Gastroenterology. 2021 Jul;161(1):33-35. doi: 10.1053/j.gastro.2021.03.046. Epub 2021 Mar 31. Gastroenterology. 2021. PMID: 33798528 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
