

Second Report of Chronic Granulomatous Disease in Jordan: Clinical and Genetic Description of 31 Patients From 21 Different Families, Including Families From Lybia and Iraq
- PMID: 33746979
- PMCID: PMC7973097
- DOI: 10.3389/fimmu.2021.639226
Second Report of Chronic Granulomatous Disease in Jordan: Clinical and Genetic Description of 31 Patients From 21 Different Families, Including Families From Lybia and Iraq
Abstract
Chronic granulomatous Disease (CGD) is a rare innate immunodeficiency disorder caused by mutations in one of the six genes (CYBA, CYBB, NCF1, NCF2, NCF4, and CYBC1/EROS) encoding the superoxide-producing nicotinamide adenine dinucleotide phosphate (NADPH)-oxidase complex in phagocytes. In the Western population, the most prevalent form of CGD (about two-thirds of all cases) is the X-linked form (X-CGD) caused by mutations in CYBB. The autosomal recessive forms (AR-CGD), due to mutations in the other genes, collectively account for the remaining one-third of CGD cases. We investigated the clinical and molecular features of 22 Jordanian, 7 Libyan, and 2 Iraqi CGD patients from 21 different families. In addition, 11 sibling patients from these families were suspected to have been died from CGD as suggested by their familial and clinical history. All patients except 9 were children of consanguineous parents. Most of the patients suffered from AR-CGD, with mutations in CYBA, NCF1, and NCF2, encoding p22 phox , p47 phox , and p67 phox proteins, respectively. AR-CGD was the most frequent form, in Jordan probably because consanguineous marriages are common in this country. Only one patient from non-consanguineous parents suffered from an X910 CGD subtype (0 indicates no protein expression). AR670 CGD and AR220 CGD appeared to be the most frequently found sub-types but also the most severe clinical forms compared to AR470 CGD. As a geographical clustering of 11 patients from eight Jordanian families exhibited the c.1171_1175delAAGCT mutation in NCF2, segregation analysis with nine polymorphic markers overlapping NCF2 indicates that a common ancestor has arisen ~1,075 years ago.
Keywords: Jordan; NADPH oxidase; autosomal recessive; chronic granulomatous disease; founder mutation; innate immunodeficiency.
Copyright © 2021 Bakri, Mollin, Beaumel, Vigne, Roux-Buisson, Al-Wahadneh, Alzyoud, Hayajneh, Daoud, Shukair, Karadshe, Sarhan, Al-Ramahi, Fauré, Rendu and Stasia.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
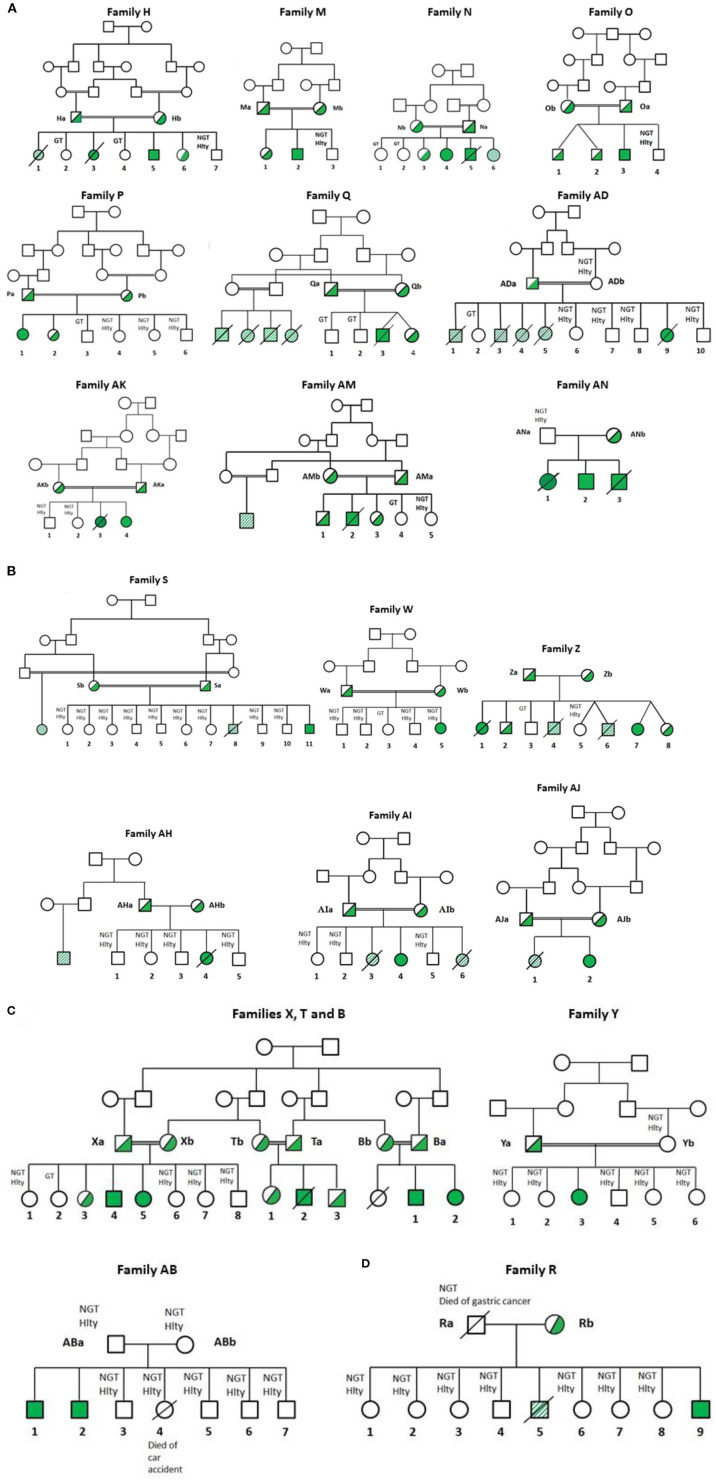
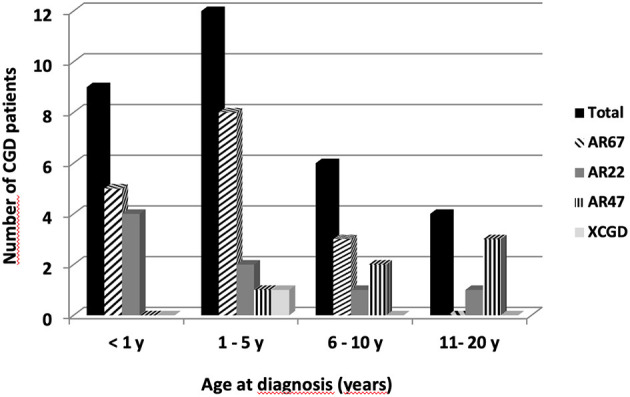
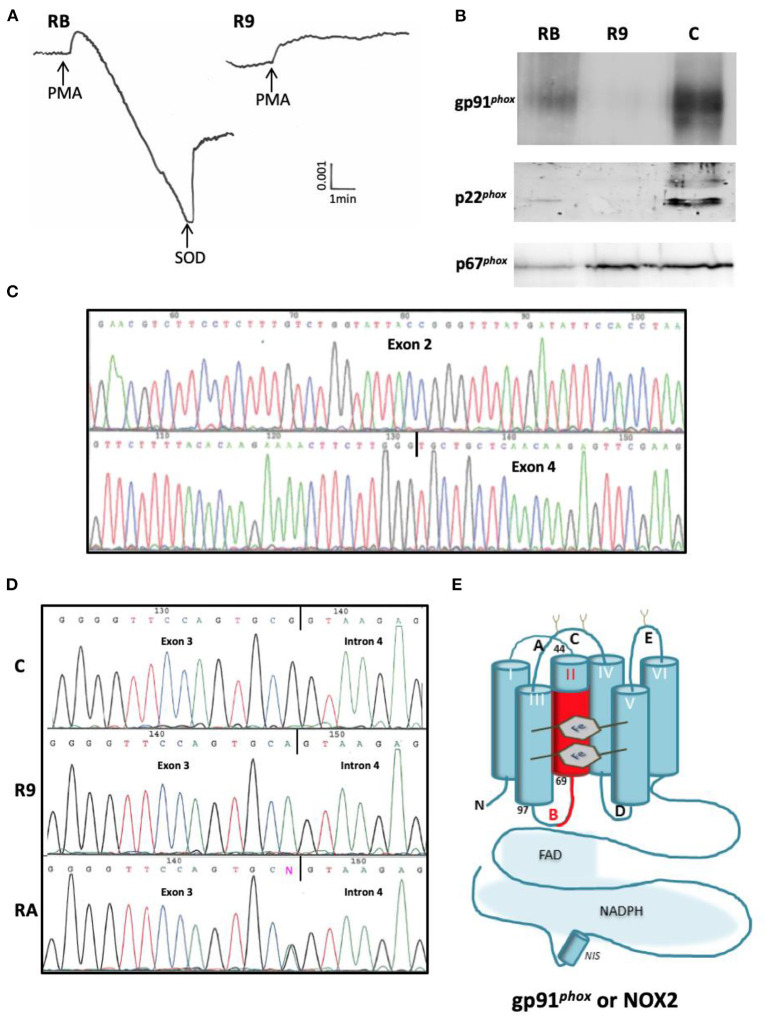
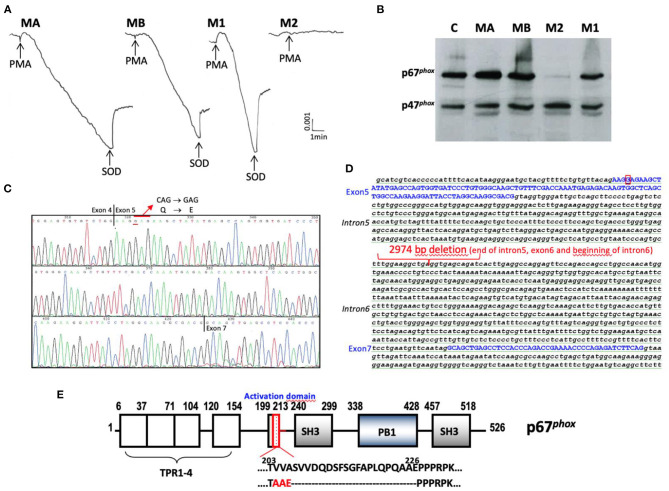
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous