

On the Wrong Track: Alterations of Ciliary Transport in Inherited Retinal Dystrophies
- PMID: 33748110
- PMCID: PMC7973215
- DOI: 10.3389/fcell.2021.623734
On the Wrong Track: Alterations of Ciliary Transport in Inherited Retinal Dystrophies
Abstract
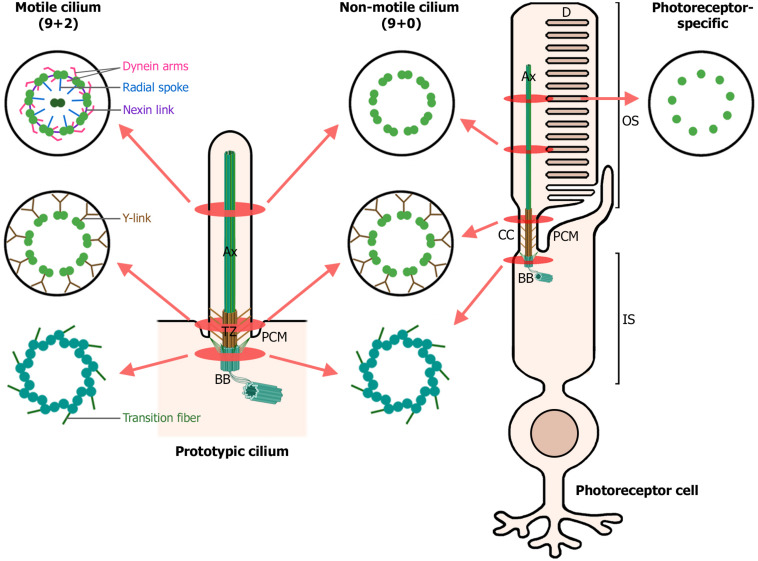
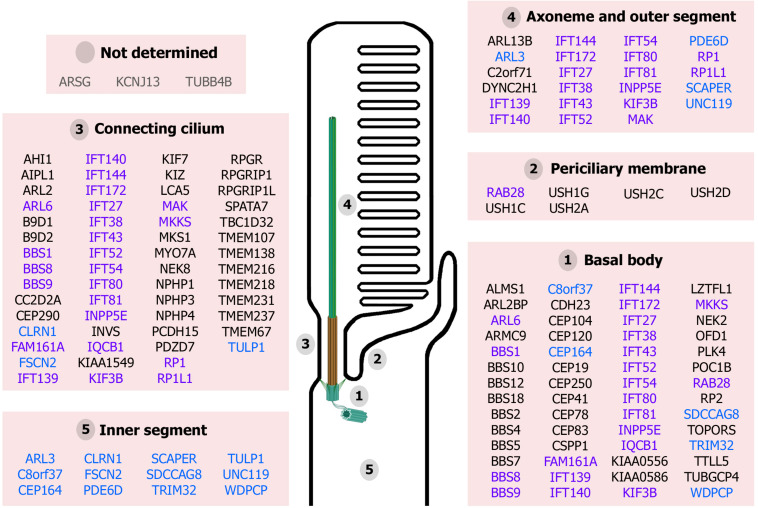
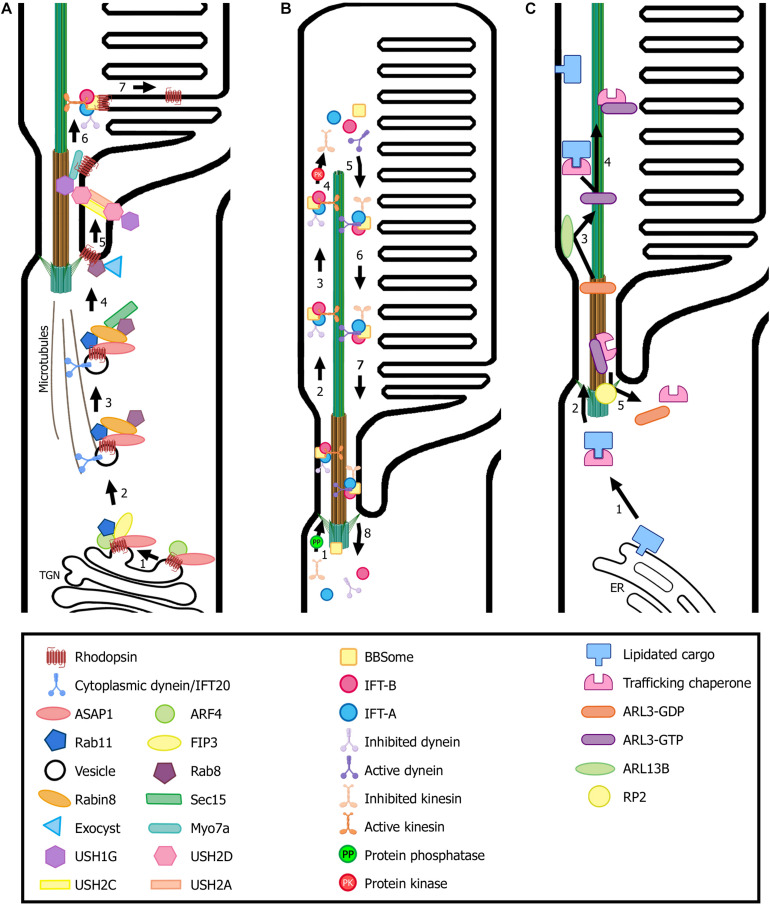
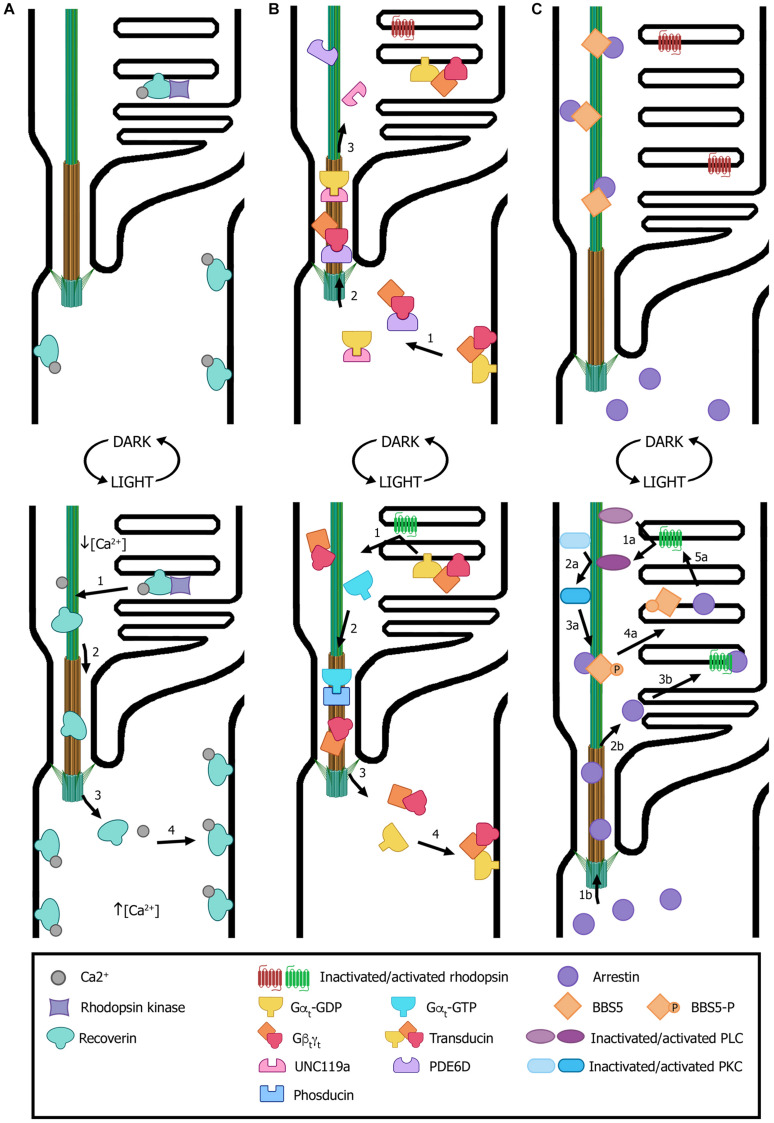
Ciliopathies are a group of heterogeneous inherited disorders associated with dysfunction of the cilium, a ubiquitous microtubule-based organelle involved in a broad range of cellular functions. Most ciliopathies are syndromic, since several organs whose cells produce a cilium, such as the retina, cochlea or kidney, are affected by mutations in ciliary-related genes. In the retina, photoreceptor cells present a highly specialized neurosensory cilium, the outer segment, stacked with membranous disks where photoreception and phototransduction occurs. The daily renewal of the more distal disks is a unique characteristic of photoreceptor outer segments, resulting in an elevated protein demand. All components necessary for outer segment formation, maintenance and function have to be transported from the photoreceptor inner segment, where synthesis occurs, to the cilium. Therefore, efficient transport of selected proteins is critical for photoreceptor ciliogenesis and function, and any alteration in either cargo delivery to the cilium or intraciliary trafficking compromises photoreceptor survival and leads to retinal degeneration. To date, mutations in more than 100 ciliary genes have been associated with retinal dystrophies, accounting for almost 25% of these inherited rare diseases. Interestingly, not all mutations in ciliary genes that cause retinal degeneration are also involved in pleiotropic pathologies in other ciliated organs. Depending on the mutation, the same gene can cause syndromic or non-syndromic retinopathies, thus emphasizing the highly refined specialization of the photoreceptor neurosensory cilia, and raising the possibility of photoreceptor-specific molecular mechanisms underlying common ciliary functions such as ciliary transport. In this review, we will focus on ciliary transport in photoreceptor cells and discuss the molecular complexity underpinning retinal ciliopathies, with a special emphasis on ciliary genes that, when mutated, cause either syndromic or non-syndromic retinal ciliopathies.
Keywords: ciliary transport; ciliopathy; inherited retinal dystrophies; intraflagellar transport; photoreceptor; sensory cilium.
Copyright © 2021 Sánchez-Bellver, Toulis and Marfany.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources