

Co-fractionation/mass spectrometry to identify protein complexes
- PMID: 33748783
- PMCID: PMC7960544
- DOI: 10.1016/j.xpro.2021.100370
Co-fractionation/mass spectrometry to identify protein complexes
Abstract
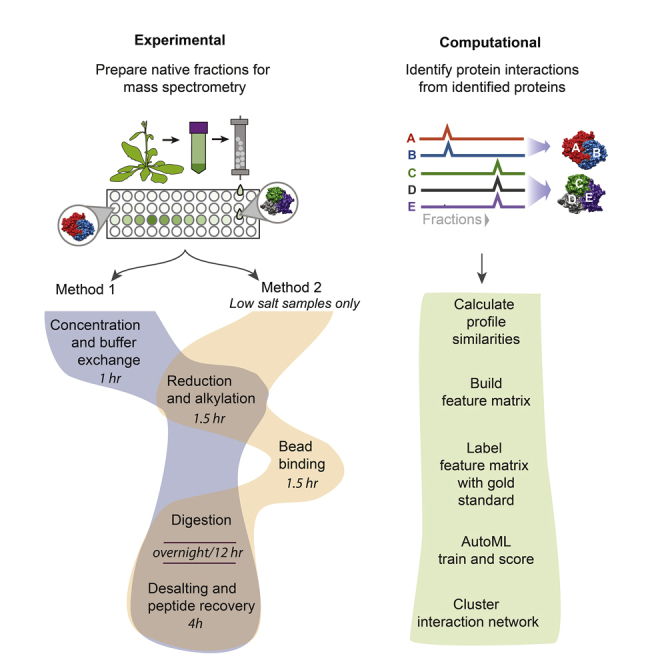
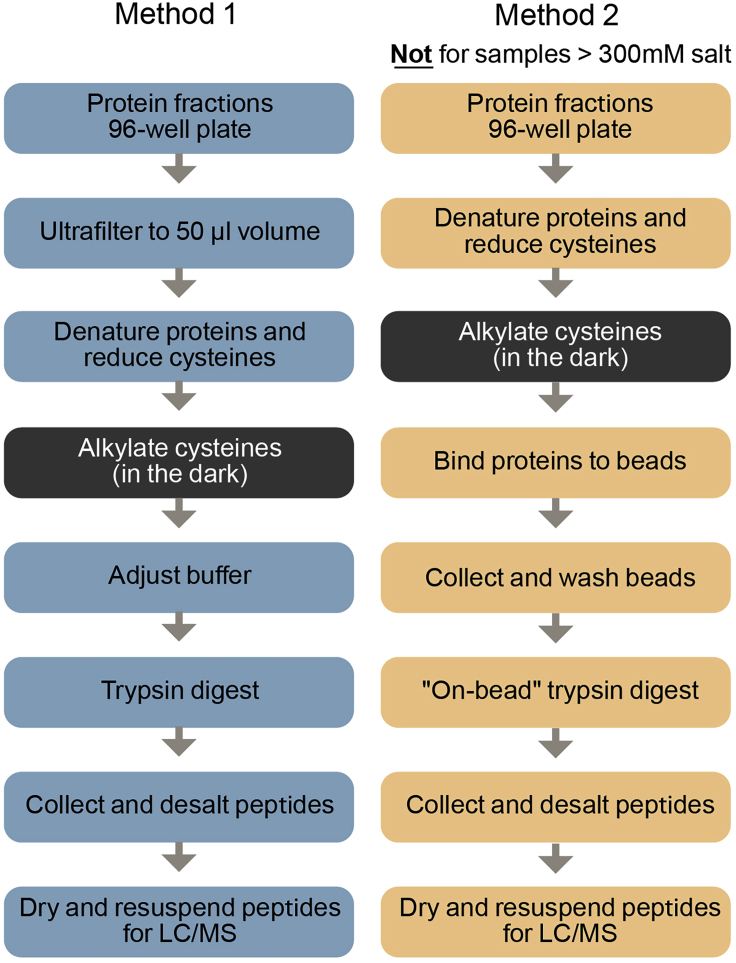
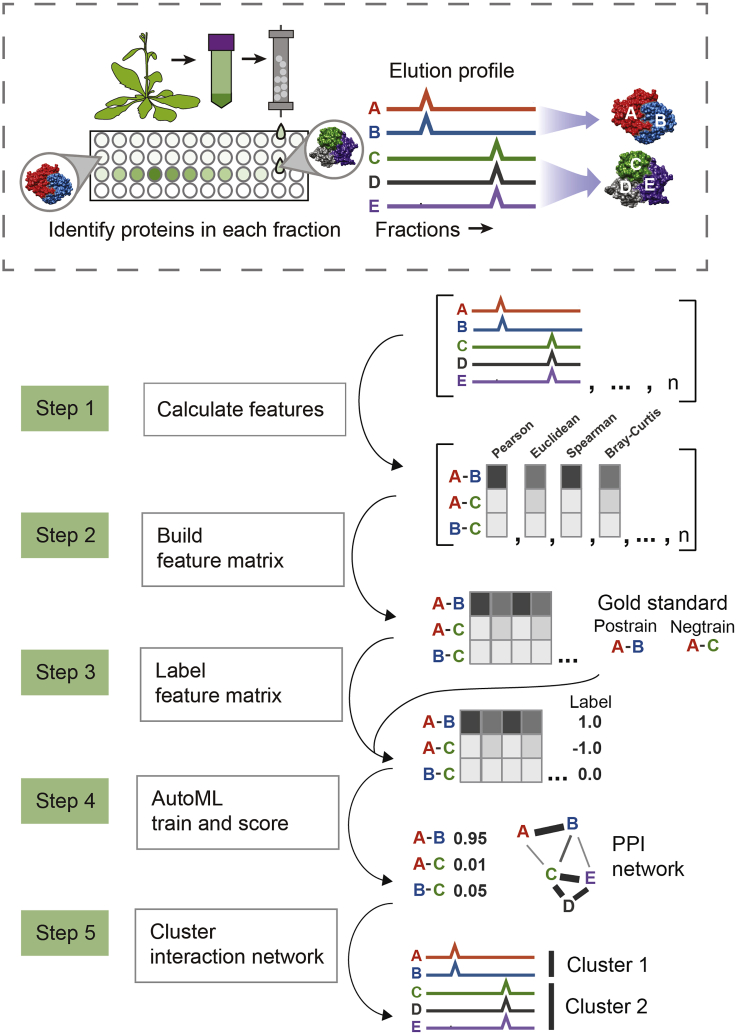
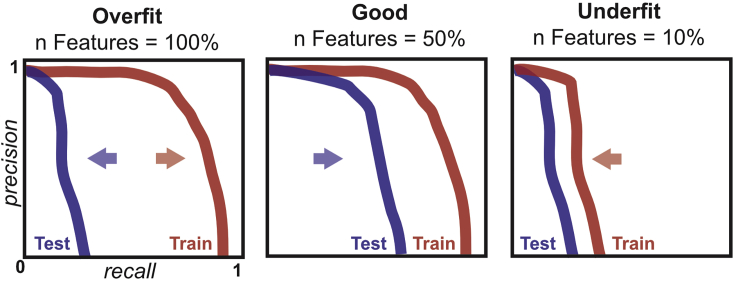
Co-fractionation/mass spectrometry (CF/MS) is a flexible and powerful method to detect physical associations of proteins. CF/MS can be applied to any tissue or organism without the need for protein-specific antibodies or epitope tags. Here, we outline two alternate protocols for MS preparation of samples (containing low or high salt) and a computational pipeline (cfmsflow) that together allow the successful application of this approach. These protocols are based on CF/MS of over 16 diverse organisms including plants and animals. For complete details on the use and execution of this protocol, please refer to McWhite et al. (2020).
Keywords: Bioinformatics; Proteomics.
© 2021 The Authors.
Conflict of interest statement
The authors have no competing interests.
Figures

References
-
- Di Tommaso P., Chatzou M., Floden E.W., Barja P.P., Palumbo E., Notredame C. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 2017;35:316–319. - PubMed
-
- Liebeskind B.J., Young R.L., Halling D.B., Aldrich R.W., Marcotte E.M. Mapping functional protein neighborhoods in the mouse brain. bioRxiv. 2020 doi: 10.1101/2020.01.26.920447. - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
