

Appyters: Turning Jupyter Notebooks into data-driven web apps
- PMID: 33748796
- PMCID: PMC7961182
- DOI: 10.1016/j.patter.2021.100213
Appyters: Turning Jupyter Notebooks into data-driven web apps
Abstract
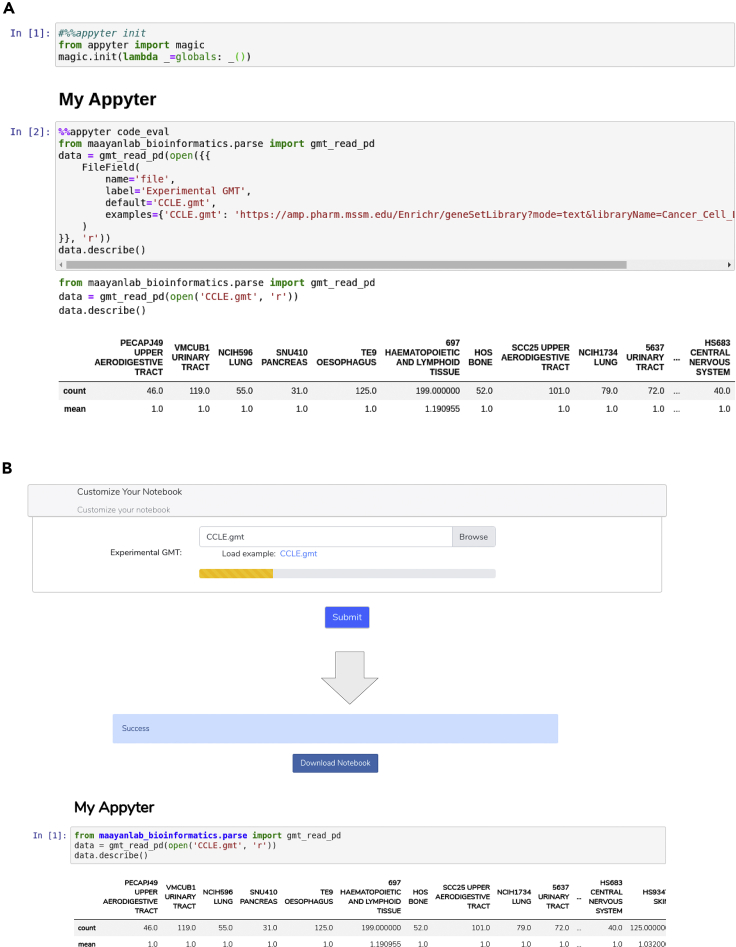
Jupyter Notebooks have transformed the communication of data analysis pipelines by facilitating a modular structure that brings together code, markdown text, and interactive visualizations. Here, we extended Jupyter Notebooks to broaden their accessibility with Appyters. Appyters turn Jupyter Notebooks into fully functional standalone web-based bioinformatics applications. Appyters present to users an entry form enabling them to upload their data and set various parameters for a multitude of data analysis workflows. Once the form is filled, the Appyter executes the corresponding notebook in the cloud, producing the output without requiring the user to interact directly with the code. Appyters were used to create many bioinformatics web-based reusable workflows, including applications to build customized machine learning pipelines, analyze omics data, and produce publishable figures. These Appyters are served in the Appyters Catalog at https://appyters.maayanlab.cloud. In summary, Appyters enable the rapid development of interactive web-based bioinformatics applications.
Keywords: RNA-seq; TCGA; big data; data analysis; data visualization; gene set enrichment analysis; machine learning; notebooks; scRNA-seq; workflow.
© 2021 The Authors.
Conflict of interest statement
The authors declare no competing interests.
Figures





References
-
- Perkel J.M. Why Jupyter is data scientists' computational notebook of choice. Nature. 2018;563:145–147. - PubMed
-
- Lau S., Hug J. nbinteract: generate interactive web pages from Jupyter notebooks. Master’s thesis (EECS Department, University of California, Berkeley) 2018
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
