

Genomic diversity of SARS-CoV-2 during early introduction into the Baltimore-Washington metropolitan area
- PMID: 33749660
- PMCID: PMC8026189
- DOI: 10.1172/jci.insight.144350
Genomic diversity of SARS-CoV-2 during early introduction into the Baltimore-Washington metropolitan area
Abstract
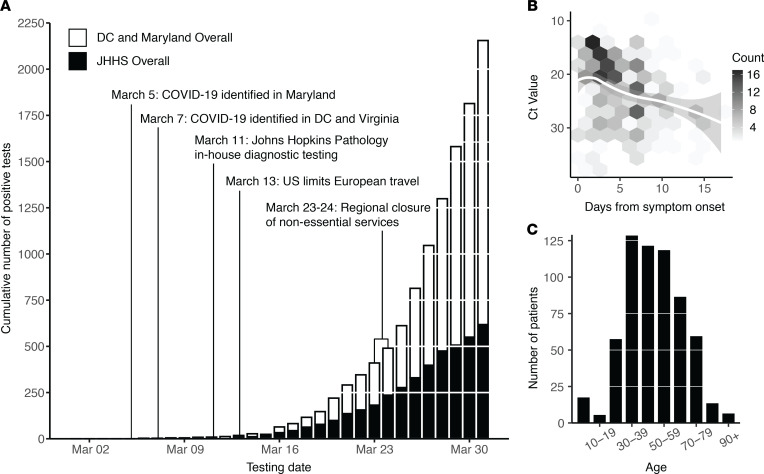
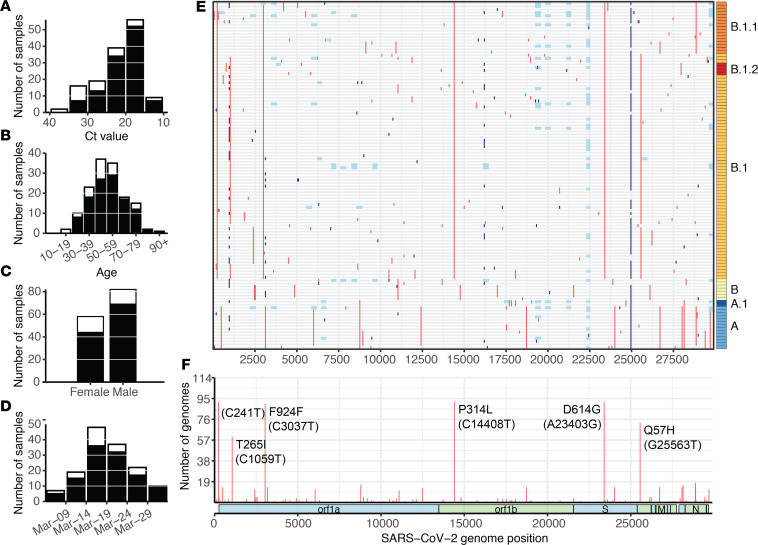
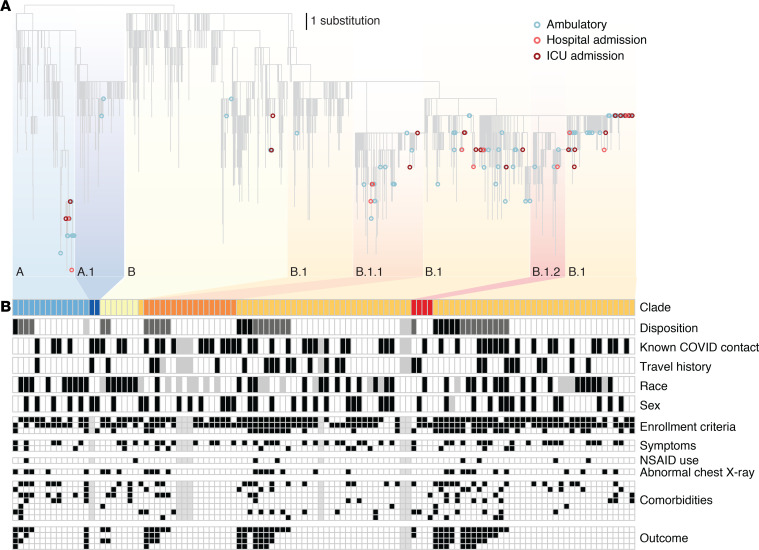
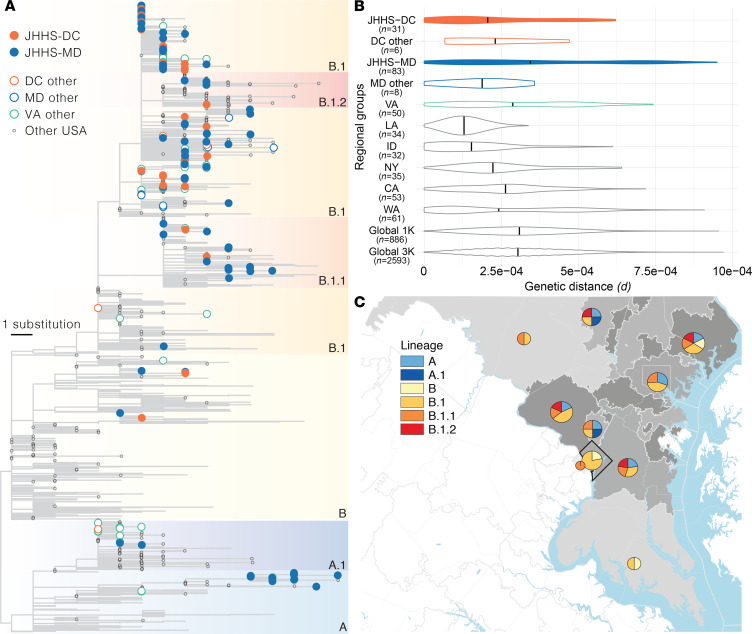
The early COVID-19 pandemic was characterized by rapid global spread. In Maryland and Washington, DC, United States, more than 2500 cases were reported within 3 weeks of the first COVID-19 detection in March 2020. We aimed to use genomic sequencing to understand the initial spread of SARS-CoV-2 - the virus that causes COVID-19 - in the region. We analyzed 620 samples collected from the Johns Hopkins Health System during March 11-31, 2020, comprising 28.6% of the total cases in Maryland and Washington, DC. From these samples, we generated 114 complete viral genomes. Analysis of these genomes alongside a subsampling of over 1000 previously published sequences showed that the diversity in this region rivaled global SARS-CoV-2 genetic diversity at that time and that the sequences belong to all of the major globally circulating lineages, suggesting multiple introductions into the region. We also analyzed these regional SARS-CoV-2 genomes alongside detailed clinical metadata and found that clinically severe cases had viral genomes belonging to all major viral lineages. We conclude that efforts to control local spread of the virus were likely confounded by the number of introductions into the region early in the epidemic and the interconnectedness of the region as a whole.
Keywords: COVID-19; Genetic variation.
Conflict of interest statement
Figures
Update of
-
Genomic Diversity of SARS-CoV-2 During Early Introduction into the United States National Capital Region.medRxiv [Preprint]. 2020 Aug 23:2020.08.13.20174136. doi: 10.1101/2020.08.13.20174136. medRxiv. 2020. Update in: JCI Insight. 2021 Mar 22;6(6):144350. doi: 10.1172/jci.insight.144350. PMID: 32817965 Free PMC article. Updated. Preprint.
References
-
- World Health Organization. Coronavirus disease 2019 (COVID-19) Situation Report – 51. https://www.who.int/docs/default-source/coronaviruse/situation-reports/2... Accessed February 12, 2021.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
