

The cell biology of Parkinson's disease
- PMID: 33749710
- PMCID: PMC8103423
- DOI: 10.1083/jcb.202012095
The cell biology of Parkinson's disease
Abstract
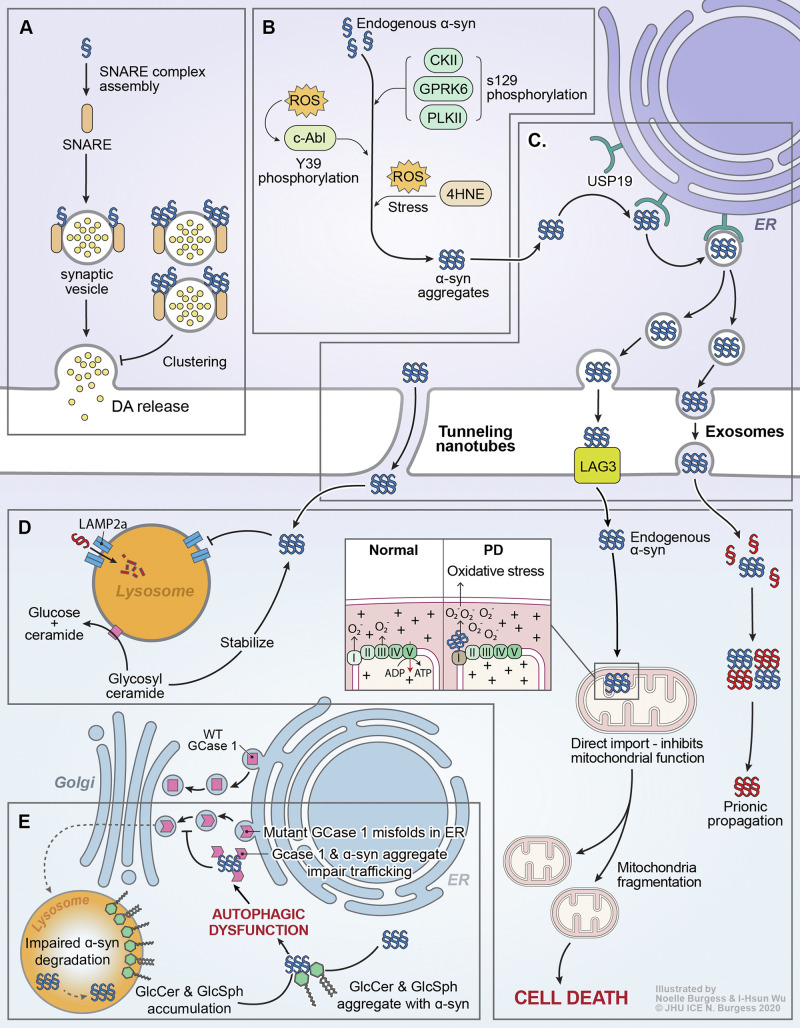
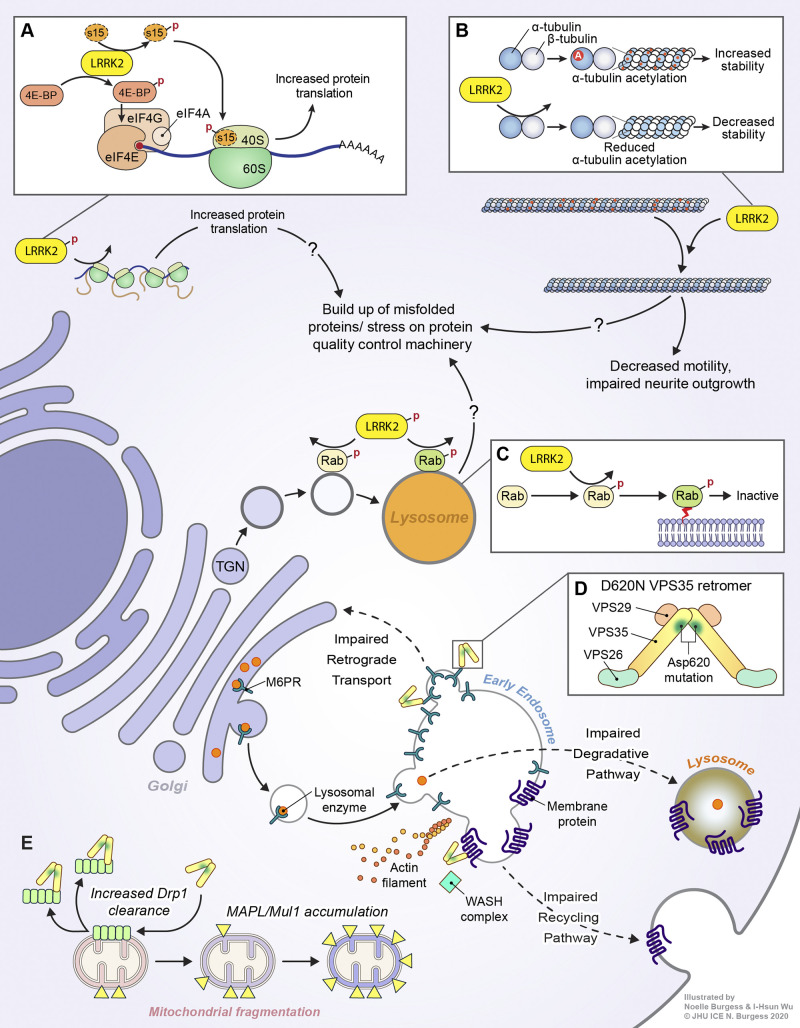
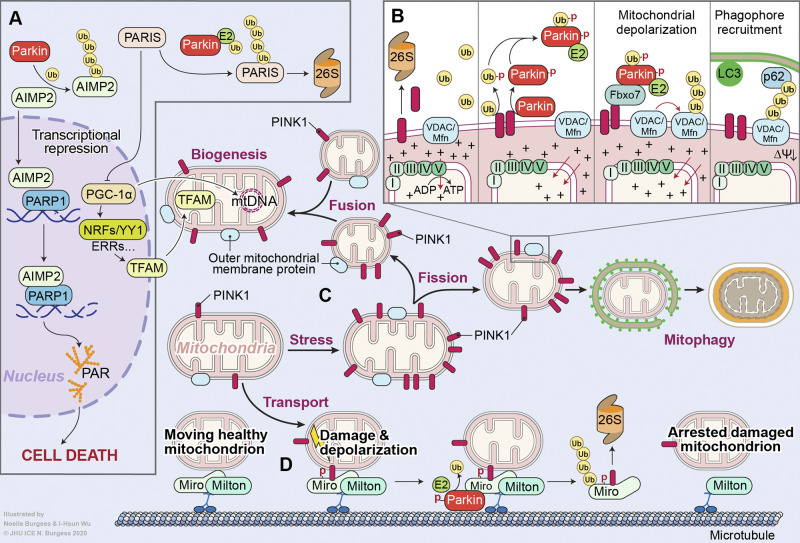
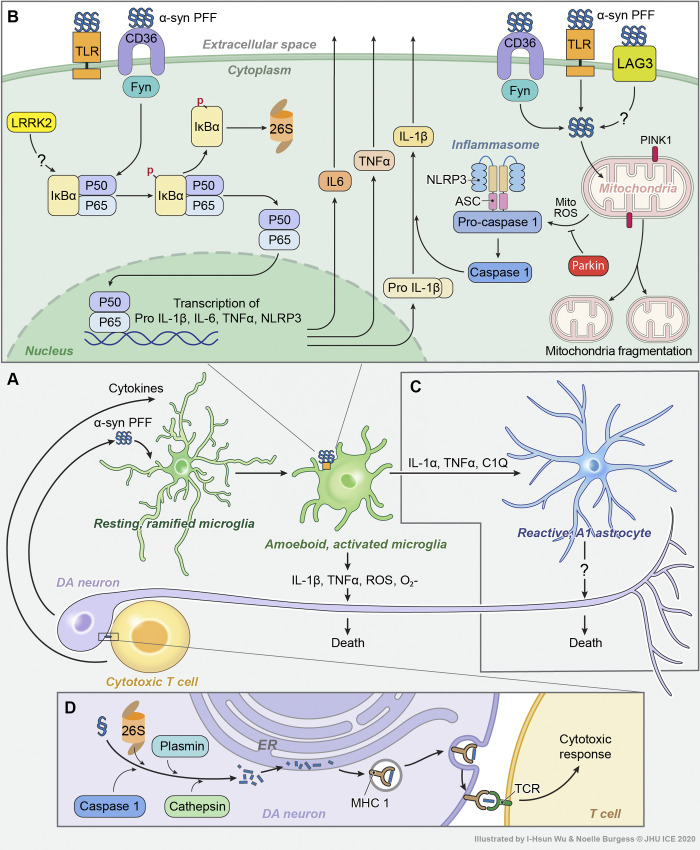
Parkinson's disease (PD) is a progressive neurodegenerative disorder resulting from the death of dopamine neurons in the substantia nigra pars compacta. Our understanding of PD biology has been enriched by the identification of genes involved in its rare, inheritable forms, termed PARK genes. These genes encode proteins including α-syn, LRRK2, VPS35, parkin, PINK1, and DJ1, which can cause monogenetic PD when mutated. Investigating the cellular functions of these proteins has been instrumental in identifying signaling pathways that mediate pathology in PD and neuroprotective mechanisms active during homeostatic and pathological conditions. It is now evident that many PD-associated proteins perform multiple functions in PD-associated signaling pathways in neurons. Furthermore, several PARK proteins contribute to non-cell-autonomous mechanisms of neuron death, such as neuroinflammation. A comprehensive understanding of cell-autonomous and non-cell-autonomous pathways involved in PD is essential for developing therapeutics that may slow or halt its progression.
© 2021 Panicker et al. This article is distributed under the terms of an Attribution–Noncommercial–Share Alike–No Mirror Sites license for the first six months after the publication date (see http://www.rupress.org/terms/). After six months it is available under a Creative Commons License (Attribution–Noncommercial–Share Alike 4.0 International license, as described at https://creativecommons.org/licenses/by-nc-sa/4.0/).
Figures
References
-
- Alegre-Abarrategui, J., Christian H., Lufino M.M., Mutihac R., Venda L.L., Ansorge O., and Wade-Martins R.. 2009. LRRK2 regulates autophagic activity and localizes to specific membrane microdomains in a novel human genomic reporter cellular model. Hum. Mol. Genet. 18:4022–4034. 10.1093/hmg/ddp346 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
