

Comprehensive single-cell sequencing reveals the stromal dynamics and tumor-specific characteristics in the microenvironment of nasopharyngeal carcinoma
- PMID: 33750785
- PMCID: PMC7943808
- DOI: 10.1038/s41467-021-21795-z
Comprehensive single-cell sequencing reveals the stromal dynamics and tumor-specific characteristics in the microenvironment of nasopharyngeal carcinoma
Abstract
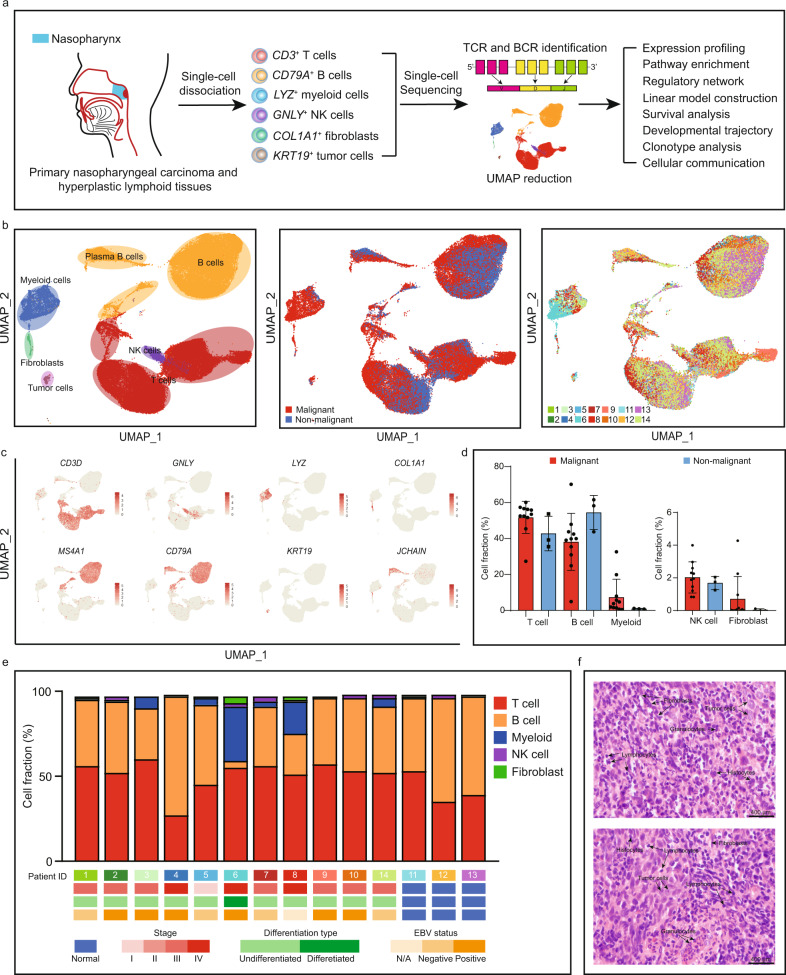
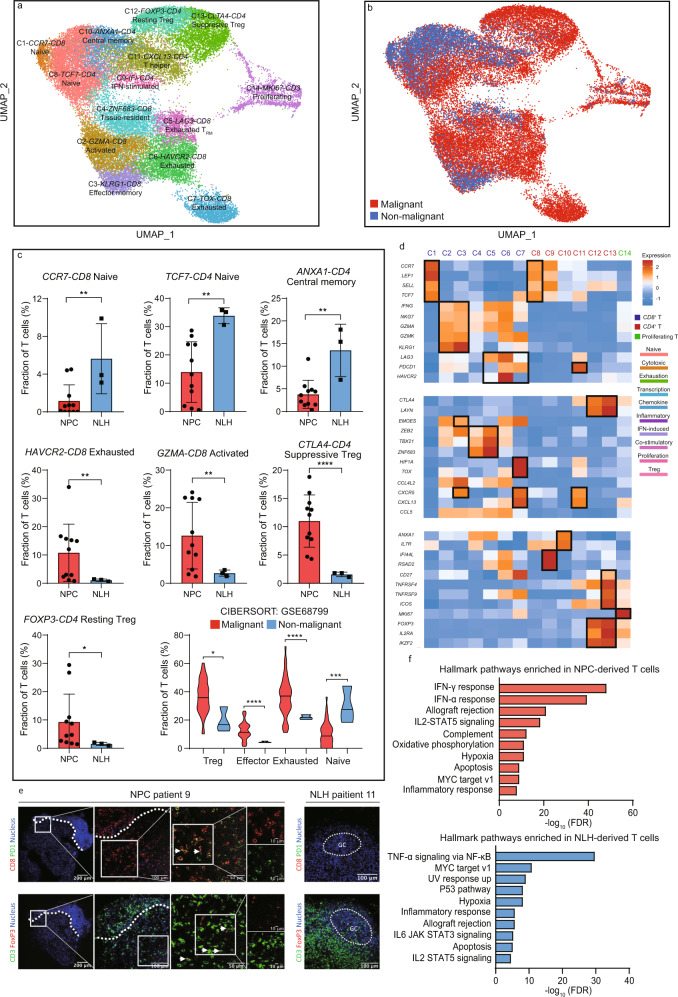
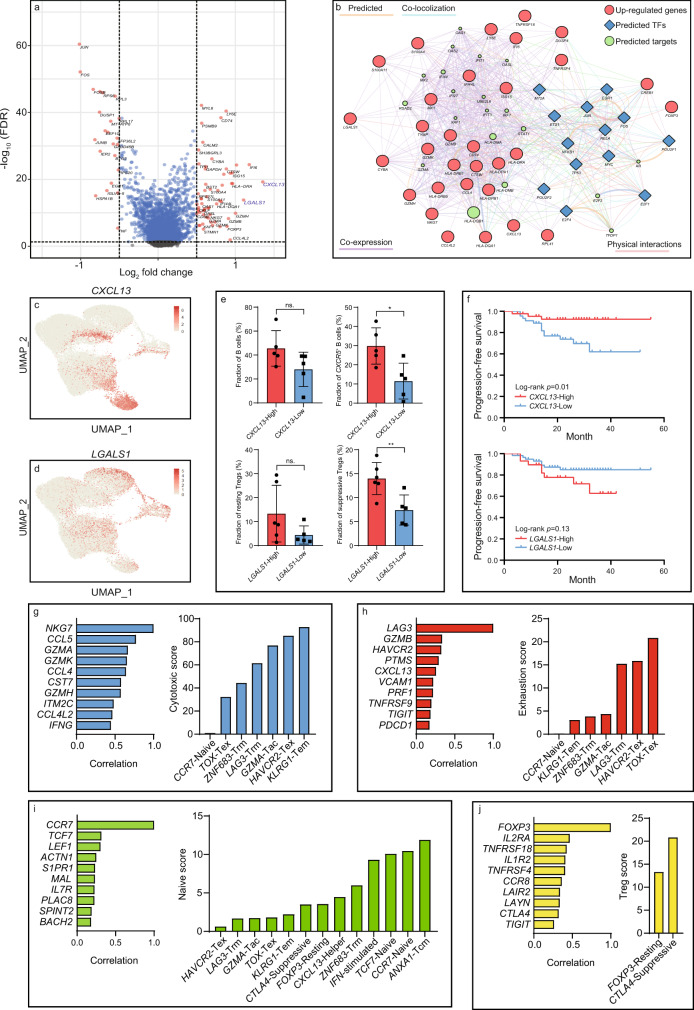
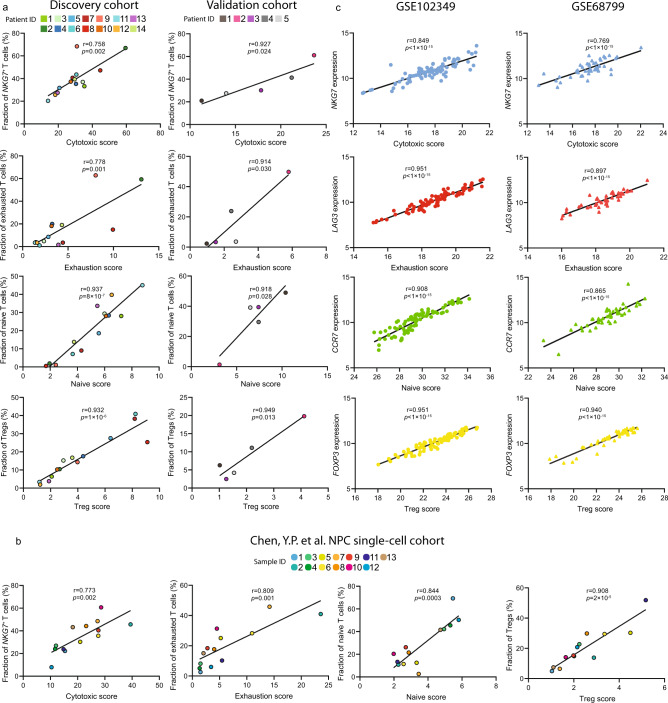
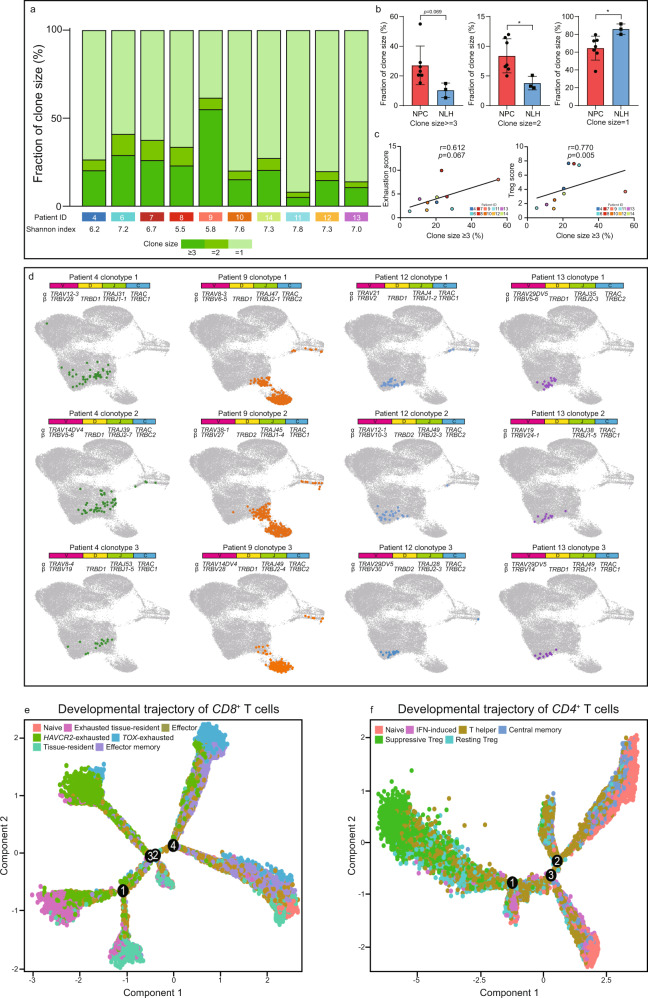
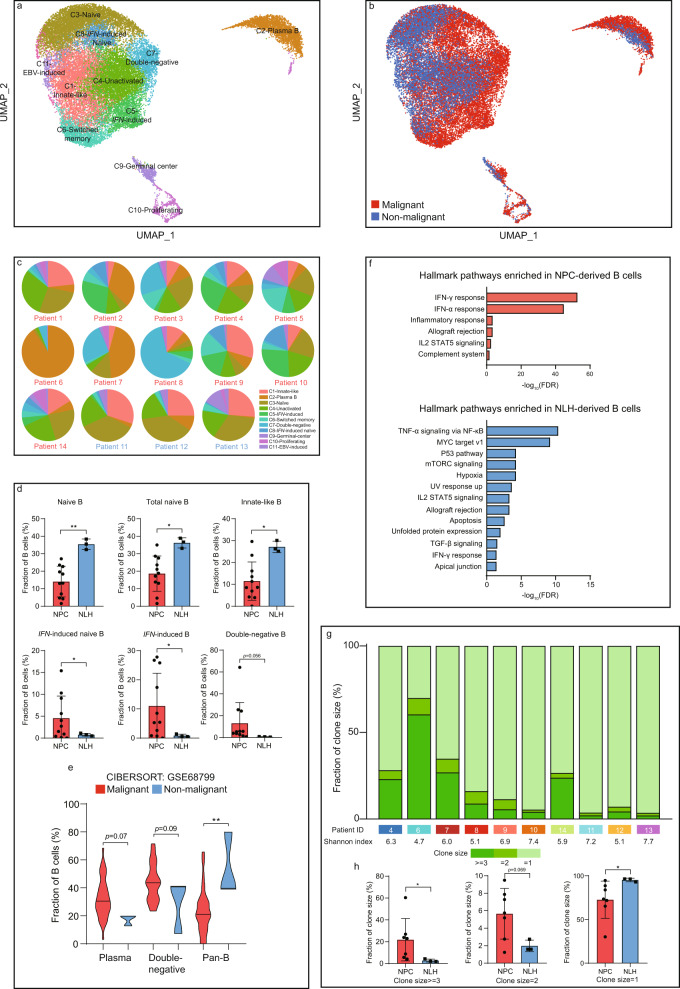
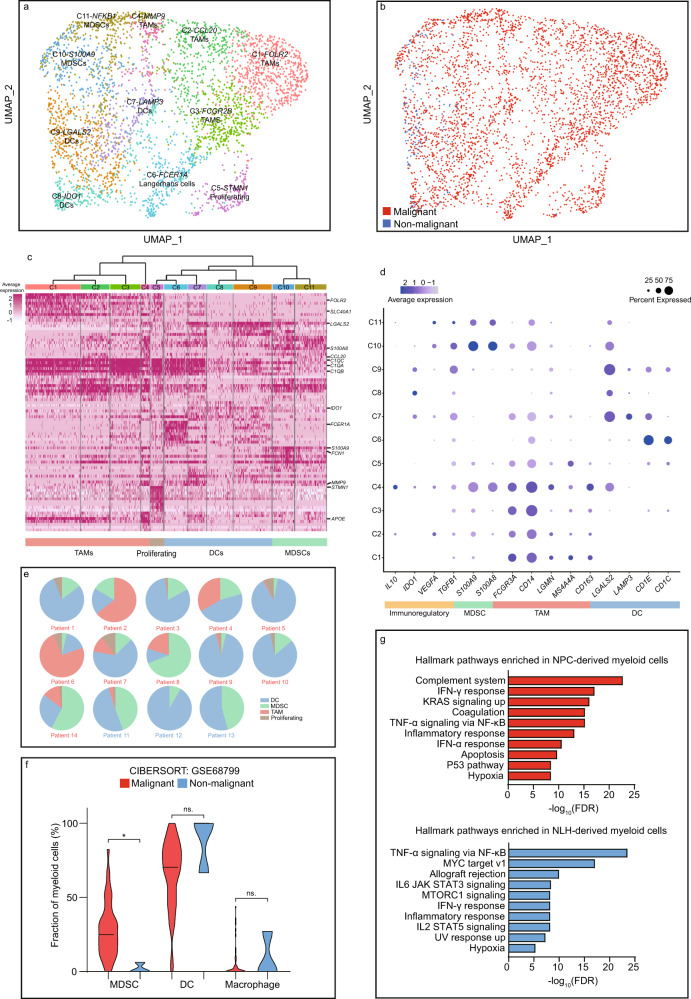
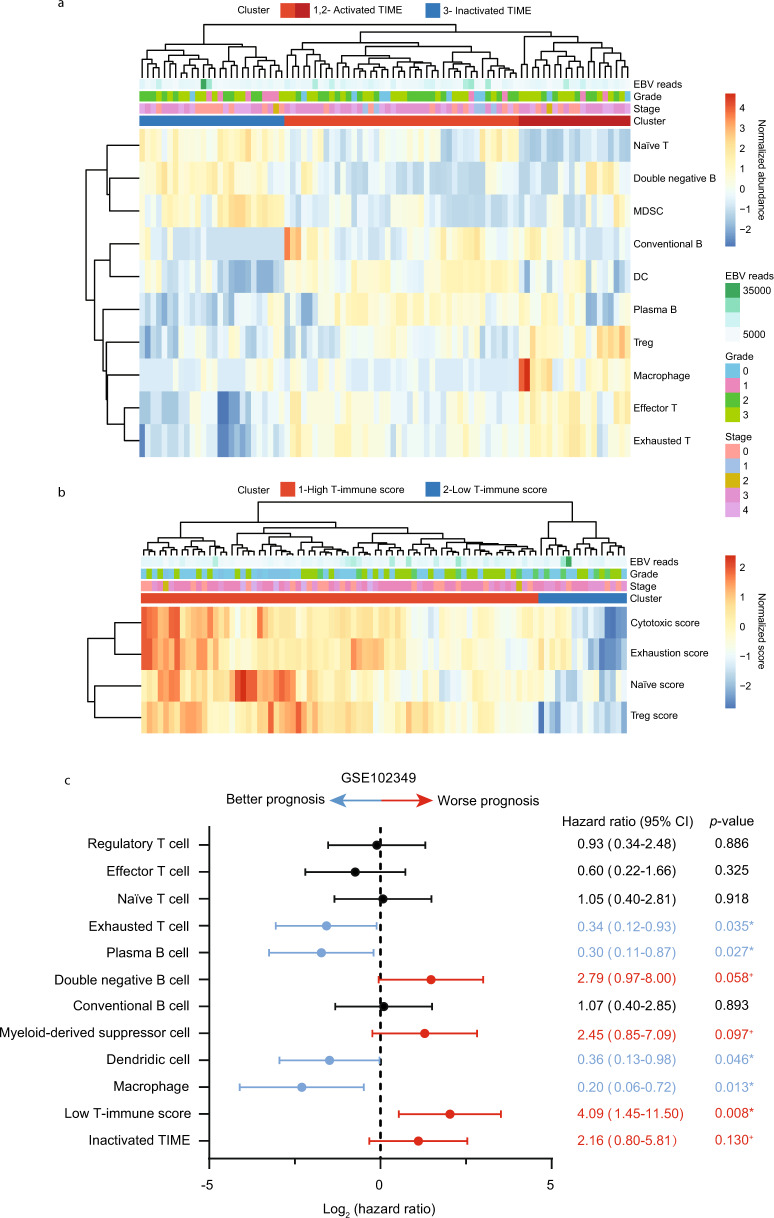
The tumor microenvironment (TME) of nasopharyngeal carcinoma (NPC) harbors a heterogeneous and dynamic stromal population. A comprehensive understanding of this tumor-specific ecosystem is necessary to enhance cancer diagnosis, therapeutics, and prognosis. However, recent advances based on bulk RNA sequencing remain insufficient to construct an in-depth landscape of infiltrating stromal cells in NPC. Here we apply single-cell RNA sequencing to 66,627 cells from 14 patients, integrated with clonotype identification on T and B cells. We identify and characterize five major stromal clusters and 36 distinct subpopulations based on genetic profiling. By comparing with the infiltrating cells in the non-malignant microenvironment, we report highly representative features in the TME, including phenotypic abundance, genetic alternations, immune dynamics, clonal expansion, developmental trajectory, and molecular interactions that profoundly influence patient prognosis and therapeutic outcome. The key findings are further independently validated in two single-cell RNA sequencing cohorts and two bulk RNA-sequencing cohorts. In the present study, we reveal the correlation between NPC-specific characteristics and progression-free survival. Together, these data facilitate the understanding of the stromal landscape and immune dynamics in NPC patients and provides deeper insights into the development of prognostic biomarkers and therapeutic targets in the TME.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Lee AWM, et al. A multicenter, phase 3, randomized trial of concurrent chemoradiotherapy plus adjuvant chemotherapy versus radiotherapy alone in patients with regionally advanced nasopharyngeal carcinoma: 10-year outcomes for efficacy and toxicity. Cancer. 2017;123:4147–4157. doi: 10.1002/cncr.30850. - DOI - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
