

Conjugated secondary 12α-hydroxylated bile acids promote liver fibrogenesis
- PMID: 33752128
- PMCID: PMC8010625
- DOI: 10.1016/j.ebiom.2021.103290
Conjugated secondary 12α-hydroxylated bile acids promote liver fibrogenesis
Abstract
Background: Significantly elevated serum and hepatic bile acid (BA) concentrations have been known to occur in patients with liver fibrosis. However, the roles of different BA species in liver fibrogenesis are not fully understood.
Methods: We quantitatively measured blood BA concentrations in nonalcoholic steatohepatitis (NASH) patients with liver fibrosis and healthy controls. We characterized BA composition in three mouse models induced by carbon tetrachloride (CCl4), streptozotocin-high fat diet (STZ-HFD), and long term HFD, respectively. The molecular mechanisms underlying the fibrosis-promoting effects of BAs were investigated in cell line models, a 3D co-culture system, and a Tgr5 (HSC-specific) KO mouse model.
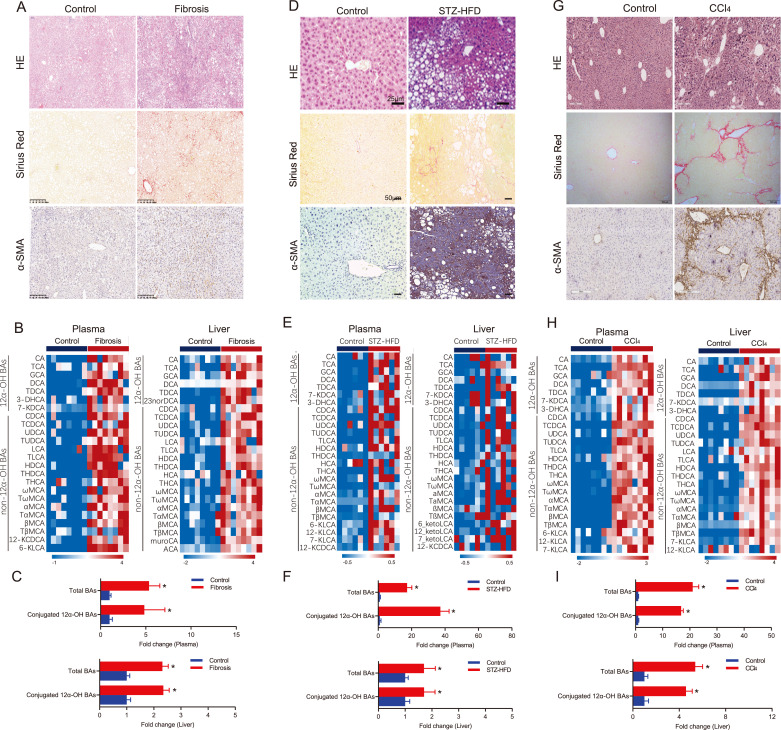
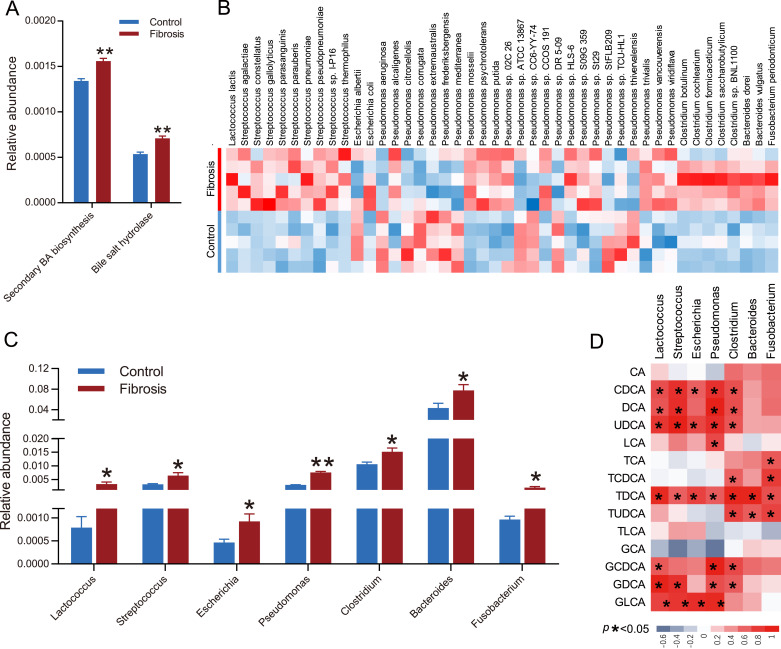
Findings: We found that a group of conjugated 12α-hydroxylated (12α-OH) BAs, such as taurodeoxycholate (TDCA) and glycodeoxycholate (GDCA), significantly increased in NASH patients and liver fibrosis mouse models. 12α-OH BAs significantly increased HSC proliferation and protein expression of fibrosis-related markers. Administration of TDCA and GDCA directly activated HSCs and promoted liver fibrogenesis in mouse models. Blockade of BA binding to TGR5 or inhibition of ERK1/2 and p38 MAPK signaling both significantly attenuated the BA-induced fibrogenesis. Liver fibrosis was attenuated in mice with Tgr5 depletion.
Interpretation: Increased hepatic concentrations of conjugated 12α-OH BAs significantly contributed to liver fibrosis via TGR5 mediated p38MAPK and ERK1/2 signaling. Strategies to antagonize TGR5 or inhibit ERK1/2 and p38 MAPK signaling may effectively prevent or reverse liver fibrosis.
Fundings: This study was supported by the National Institutes of Health/National Cancer Institute Grant 1U01CA188387-01A1, the National Key Research and Development Program of China (2017YFC0906800); the State Key Program of National Natural Science Foundation (81430062); the National Natural Science Foundation of China (81974073, 81774196), China Postdoctoral Science Foundation funded project, China (2016T90381), and E-institutes of Shanghai Municipal Education Commission, China (E03008).
Keywords: 12α-hydroxylated bile acids; ERK1/2; G protein-coupled bile acid receptor; Hepatic stellate cell; Liver fibrosis; TGR5; p38MAPK.
Copyright © 2021 The Authors. Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
Declaration of Competing Interests The authors declare no competing interests.
Figures






References
-
- Shneider BL, Gonzalez-Peralta R, Roberts EA. Controversies in the management of pediatric liver disease: Hepatitis B, C and NAFLD: summary of a single topic conference. Hepatology. 2006;44(5):1344–1354. - PubMed
-
- Bugianesi E, Leone N, Vanni E. Expanding the natural history of nonalcoholic steatohepatitis: from cryptogenic cirrhosis to hepatocellular carcinoma. Gastroenterology. 2002;123(1):134–140. - PubMed
-
- McPherson S, Hardy T, Henderson E, Burt AD, Day CP, Anstee QM. Evidence of NAFLD progression from steatosis to fibrosing-steatohepatitis using paired biopsies: Implications for prognosis and clinical management. J Hepatol. 2015;62(5):1148–1155. - PubMed
-
- Chang TT, Liaw YF, Wu SS. Long-term entecavir therapy results in the reversal of fibrosis/cirrhosis and continued histological improvement in patients with chronic hepatitis B. Hepatology. 2010;52(3):886–893. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
