

Loss of autophagy protein ATG5 impairs cardiac capacity in mice and humans through diminishing mitochondrial abundance and disrupting Ca2+ cycling
- PMID: 33752242
- PMCID: PMC9074988
- DOI: 10.1093/cvr/cvab112
Loss of autophagy protein ATG5 impairs cardiac capacity in mice and humans through diminishing mitochondrial abundance and disrupting Ca2+ cycling
Abstract
Aims: Autophagy protects against the development of cardiac hypertrophy and failure. While aberrant Ca2+ handling promotes myocardial remodelling and contributes to contractile dysfunction, the role of autophagy in maintaining Ca2+ homeostasis remains elusive. Here, we examined whether Atg5 deficiency-mediated autophagy promotes early changes in subcellular Ca2+ handling in ventricular cardiomyocytes, and whether those alterations associate with compromised cardiac reserve capacity, which commonly precedes the onset of heart failure.
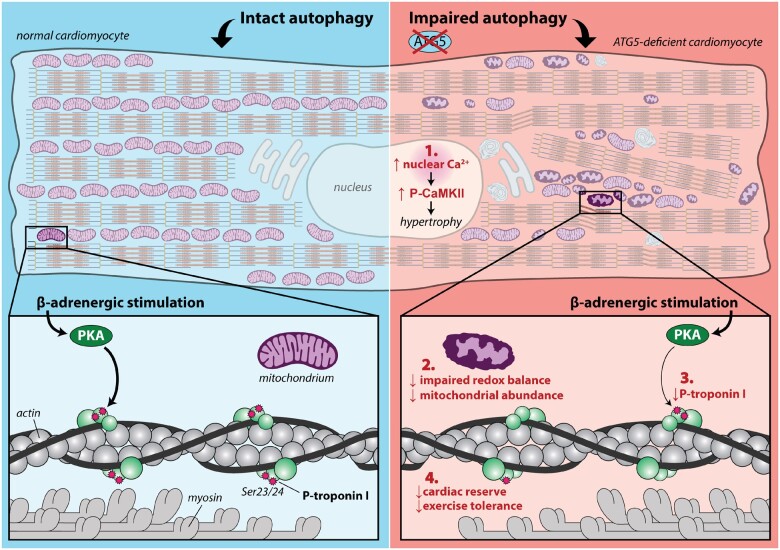
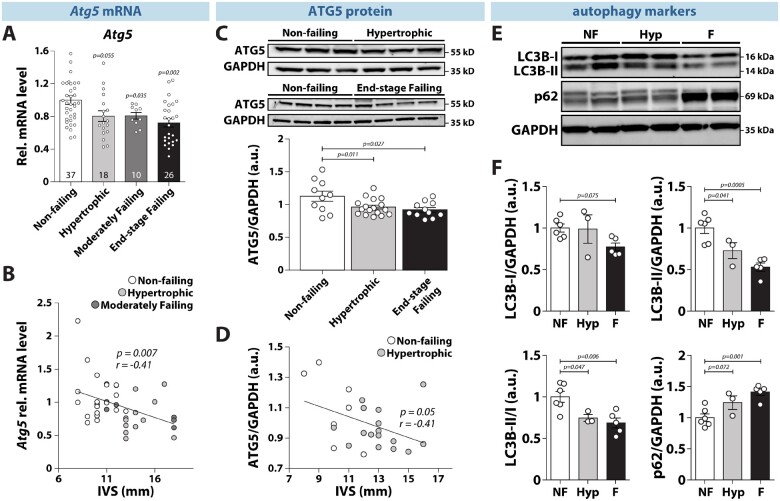
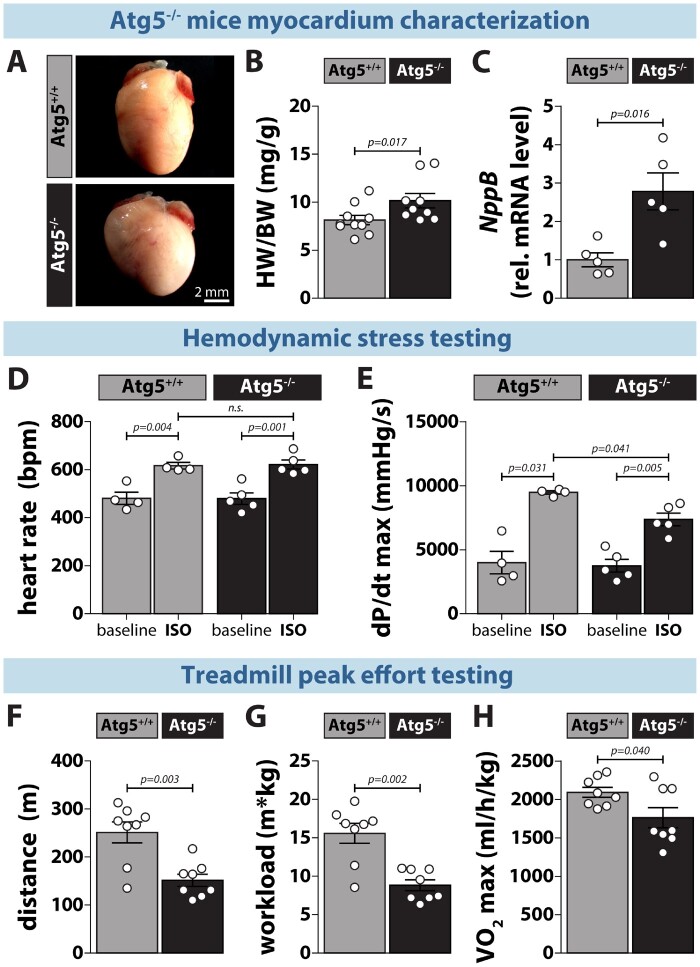
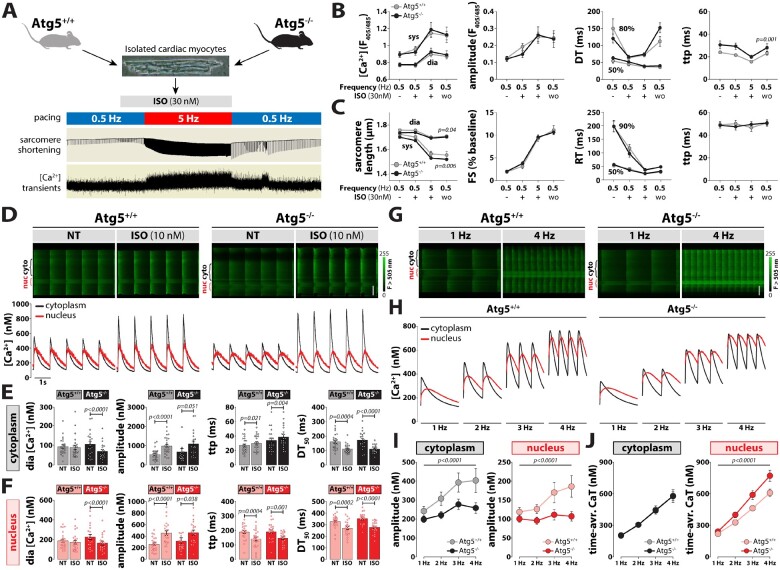
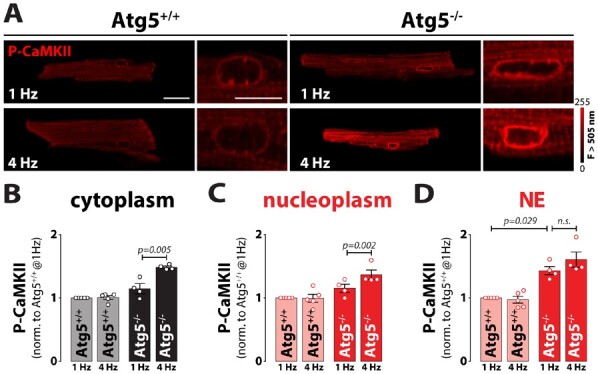
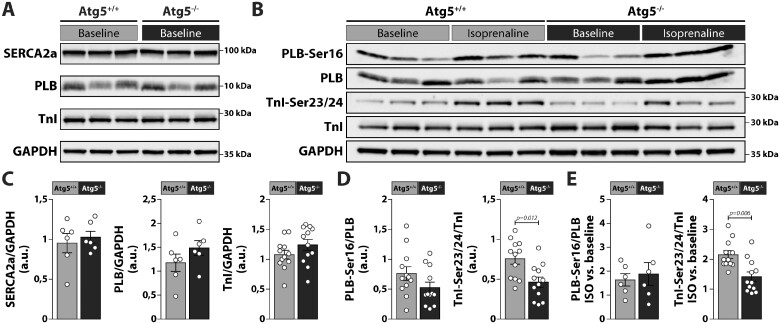
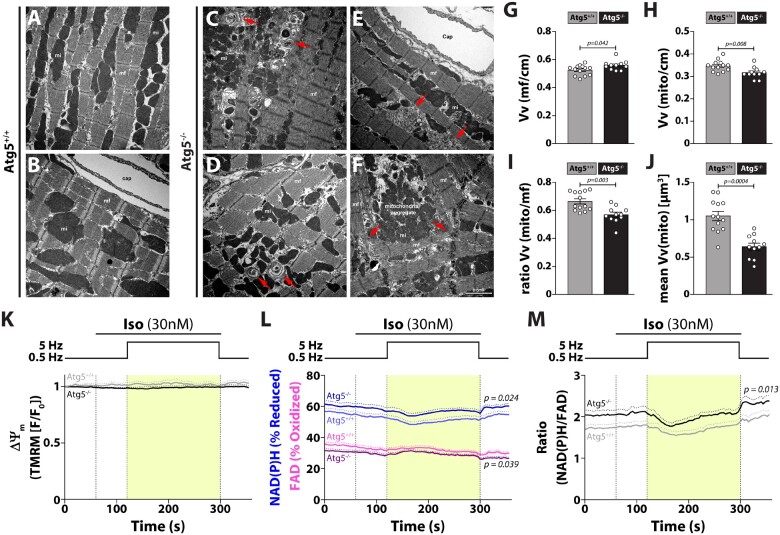
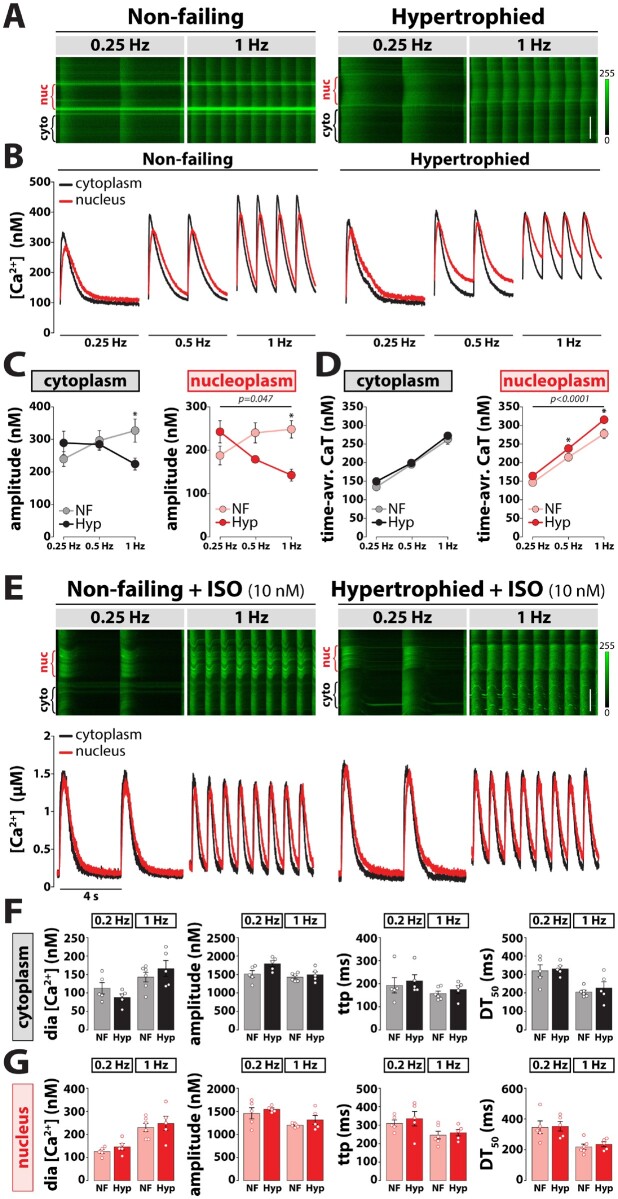
Methods and results: RT-qPCR and immunoblotting demonstrated reduced Atg5 gene and protein expression and decreased abundancy of autophagy markers in hypertrophied and failing human hearts. The function of ATG5 was examined using cardiomyocyte-specific Atg5-knockout mice (Atg5-/-). Before manifesting cardiac dysfunction, Atg5-/- mice showed compromised cardiac reserve in response to β-adrenergic stimulation. Consequently, effort intolerance and maximal oxygen consumption were reduced during treadmill-based exercise tolerance testing. Mechanistically, cellular imaging revealed that Atg5 deprivation did not alter spatial and functional organization of intracellular Ca2+ stores or affect Ca2+ cycling in response to slow pacing or upon acute isoprenaline administration. However, high-frequency stimulation exposed stunted amplitude of Ca2+ transients, augmented nucleoplasmic Ca2+ load, and increased CaMKII activity, especially in the nuclear region of hypertrophied Atg5-/- cardiomyocytes. These changes in Ca2+ cycling were recapitulated in hypertrophied human cardiomyocytes. Finally, ultrastructural analysis revealed accumulation of mitochondria with reduced volume and size distribution, meanwhile functional measurements showed impaired redox balance in Atg5-/- cardiomyocytes, implying energetic unsustainability due to overcompensation of single mitochondria, particularly under increased workload.
Conclusion: Loss of cardiac Atg5-dependent autophagy reduces mitochondrial abundance and causes subtle alterations in subcellular Ca2+ cycling upon increased workload in mice. Autophagy-related impairment of Ca2+ handling is progressively worsened by β-adrenergic signalling in ventricular cardiomyocytes, thereby leading to energetic exhaustion and compromised cardiac reserve.
Keywords: Autophagy; Beta-adrenergic signalling; Calcium; Cardiomyocytes; Mitochondria.
© The Author(s) 2021. Published by Oxford University Press on behalf of the European Society of Cardiology.
Figures
Comment in
-
Regulation of calcium handling by autophagy: a novel mechanism limiting cardiac hypertrophy and dysfunction?Cardiovasc Res. 2022 May 6;118(6):1377-1379. doi: 10.1093/cvr/cvac035. Cardiovasc Res. 2022. PMID: 35425968 No abstract available.
References
-
- Abdellatif M, Sedej S, Carmona-Gutierrez D, Madeo F, Kroemer G. Autophagy in cardiovascular aging. Circ Res 2018;123:803–824. - PubMed
-
- Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, Nishida K, Hori M, Mizushima N, Otsu K. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med 2007;13:619–624. - PubMed
-
- Eisenberg T, Abdellatif M, Schroeder S, Primessnig U, Stekovic S, Pendl T, Harger A, Schipke J, Zimmermann A, Schmidt A, Tong M, Ruckenstuhl C, Dammbrueck C, Gross AS, Herbst V, Magnes C, Trausinger G, Narath S, Meinitzer A, Hu Z, Kirsch A, Eller K, Carmona-Gutierrez D, Büttner S, Pietrocola F, Knittelfelder O, Schrepfer E, Rockenfeller P, Simonini C, Rahn A, Horsch M, Moreth K, Beckers J, Fuchs H, Gailus-Durner V, Neff F, Janik D, Rathkolb B, Rozman J, de Angelis MH, Moustafa T, Haemmerle G, Mayr M, Willeit P, von Frieling-Salewsky M, Pieske B, Scorrano L, Pieber T, Pechlaner R, Willeit J, Sigrist SJ, Linke WA, Mühlfeld C, Sadoshima J, Dengjel J, Kiechl S, Kroemer G, Sedej S, Madeo F. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat Med 2016;22:1428–1438. - PMC - PubMed
-
- Taneike M, Yamaguchi O, Nakai A, Hikoso S, Takeda T, Mizote I, Oka T, Tamai T, Oyabu J, Murakawa T, Nishida K, Shimizu T, Hori M, Komuro I, Takuji Shirasawa TS, Mizushima N, Otsu K. Inhibition of autophagy in the heart induces age-related cardiomyopathy. Autophagy 2010;6:600–606. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
