

A glutaminase isoform switch drives therapeutic resistance and disease progression of prostate cancer
- PMID: 33753479
- PMCID: PMC8020804
- DOI: 10.1073/pnas.2012748118
A glutaminase isoform switch drives therapeutic resistance and disease progression of prostate cancer
Abstract
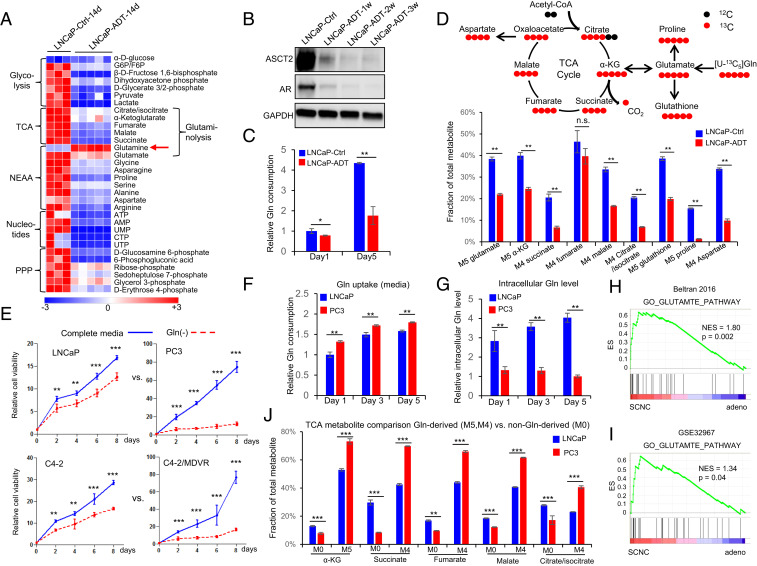
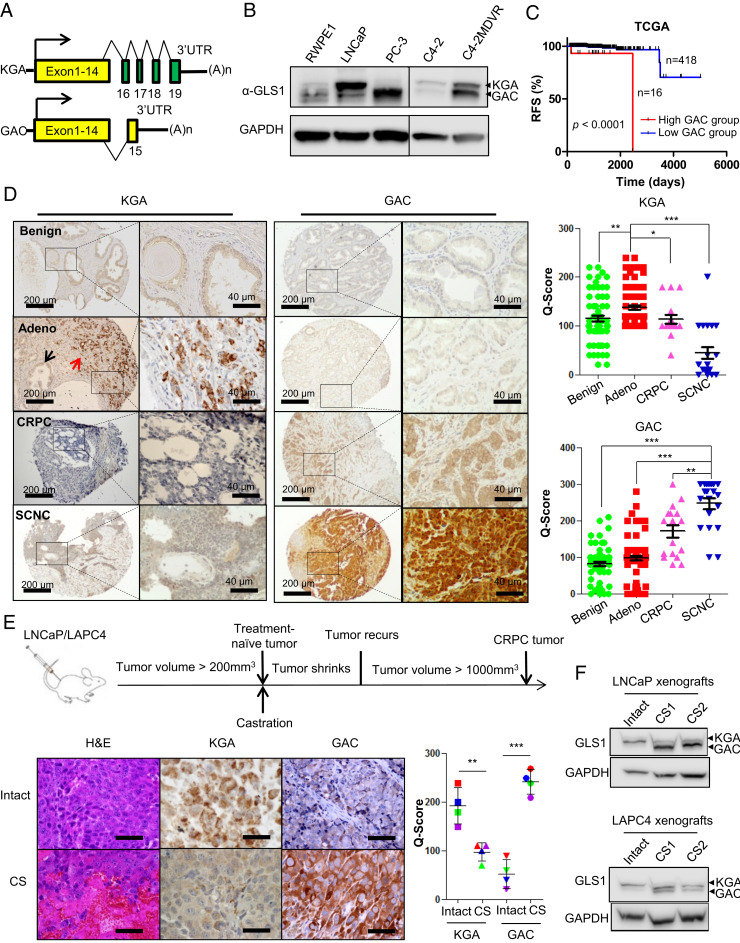
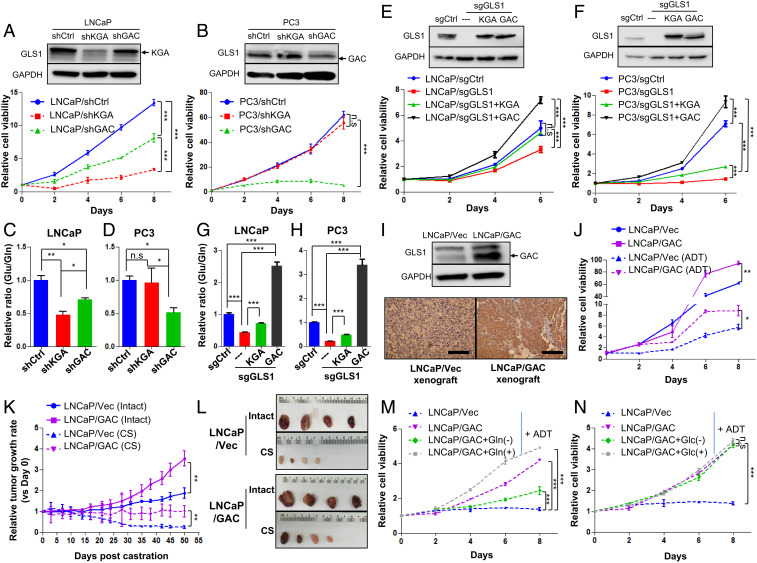
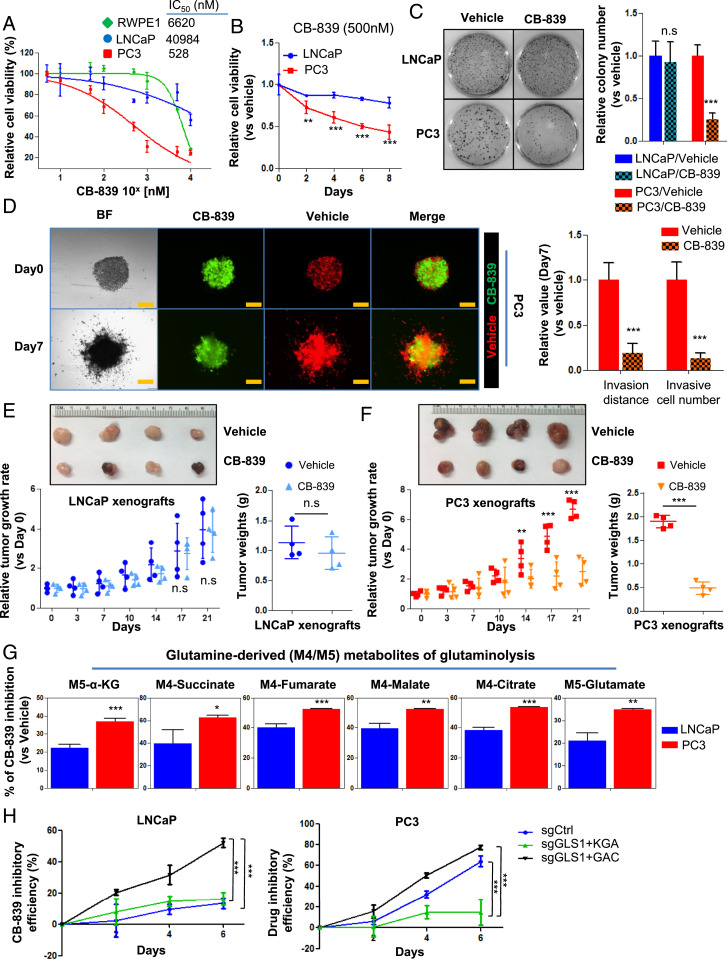
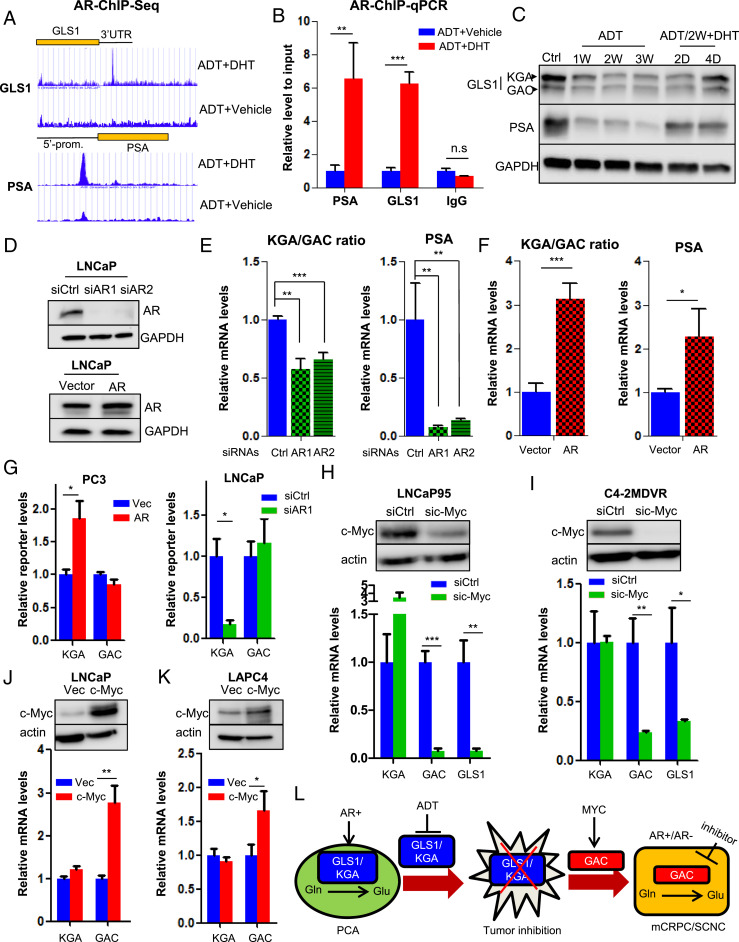
Cellular metabolism in cancer is significantly altered to support the uncontrolled tumor growth. How metabolic alterations contribute to hormonal therapy resistance and disease progression in prostate cancer (PCa) remains poorly understood. Here we report a glutaminase isoform switch mechanism that mediates the initial therapeutic effect but eventual failure of hormonal therapy of PCa. Androgen deprivation therapy inhibits the expression of kidney-type glutaminase (KGA), a splicing isoform of glutaminase 1 (GLS1) up-regulated by androgen receptor (AR), to achieve therapeutic effect by suppressing glutaminolysis. Eventually the tumor cells switch to the expression of glutaminase C (GAC), an androgen-independent GLS1 isoform with more potent enzymatic activity, under the androgen-deprived condition. This switch leads to increased glutamine utilization, hyperproliferation, and aggressive behavior of tumor cells. Pharmacological inhibition or RNA interference of GAC shows better treatment effect for castration-resistant PCa than for hormone-sensitive PCa in vitro and in vivo. In summary, we have identified a metabolic function of AR action in PCa and discovered that the GLS1 isoform switch is one of the key mechanisms in therapeutic resistance and disease progression.
Keywords: GAC; glutaminase; prostate cancer; therapeutic resistance.
Conflict of interest statement
Competing interest statement: J.H. is a consultant for or owns shares in the following companies: Kingmed, MoreHealth, OptraScan, Genetron, Omnitura, Vetonco, York Biotechnology, Genecode, and Sisu Pharma.
Figures
Comment in
-
Uro-Science.J Urol. 2021 Nov;206(5):1319-1321. doi: 10.1097/JU.0000000000002153. Epub 2021 Aug 16. J Urol. 2021. PMID: 34392697 No abstract available.
References
-
- Barbieri C. E., Rubin M. A., Molecular characterization of prostate cancer following androgen deprivation: The devil in the details. Eur. Urol. 66, 40–41 (2014). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
