

A first-generation pediatric cancer dependency map
- PMID: 33753930
- PMCID: PMC8049517
- DOI: 10.1038/s41588-021-00819-w
A first-generation pediatric cancer dependency map
Abstract
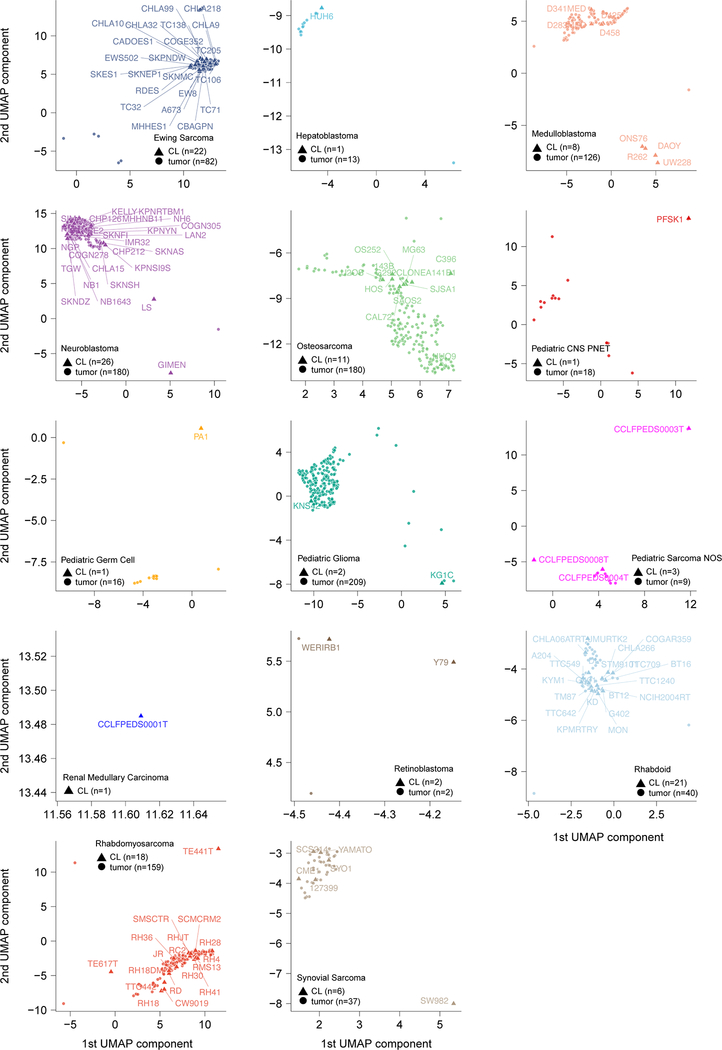
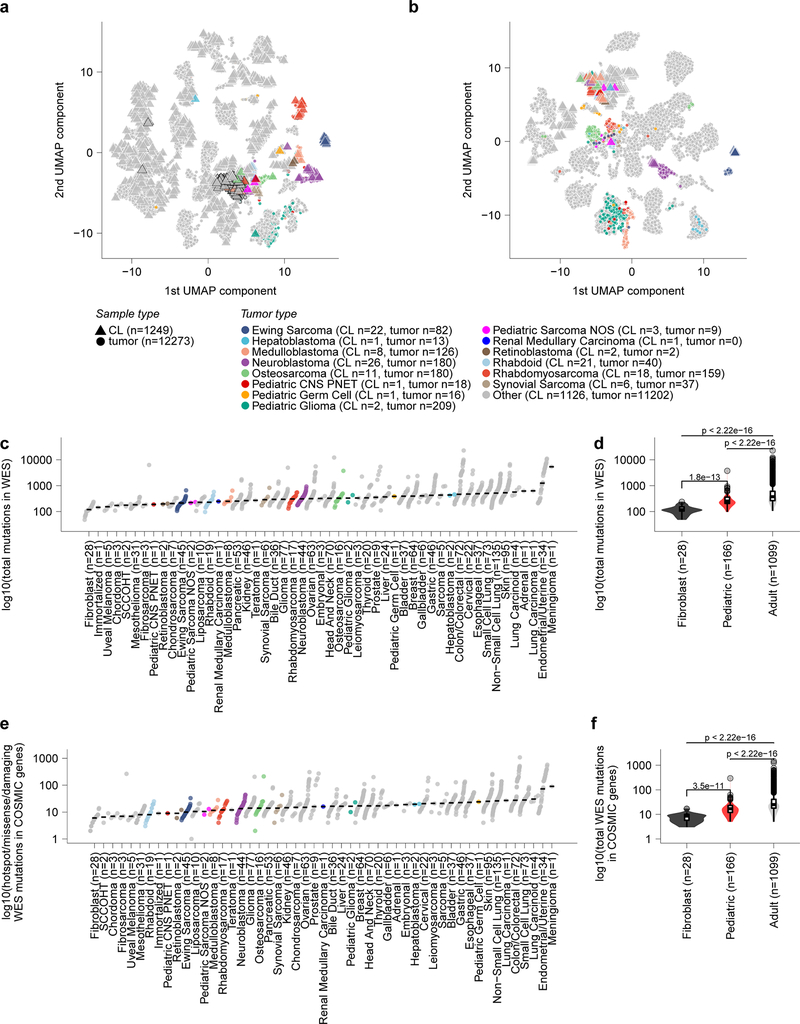
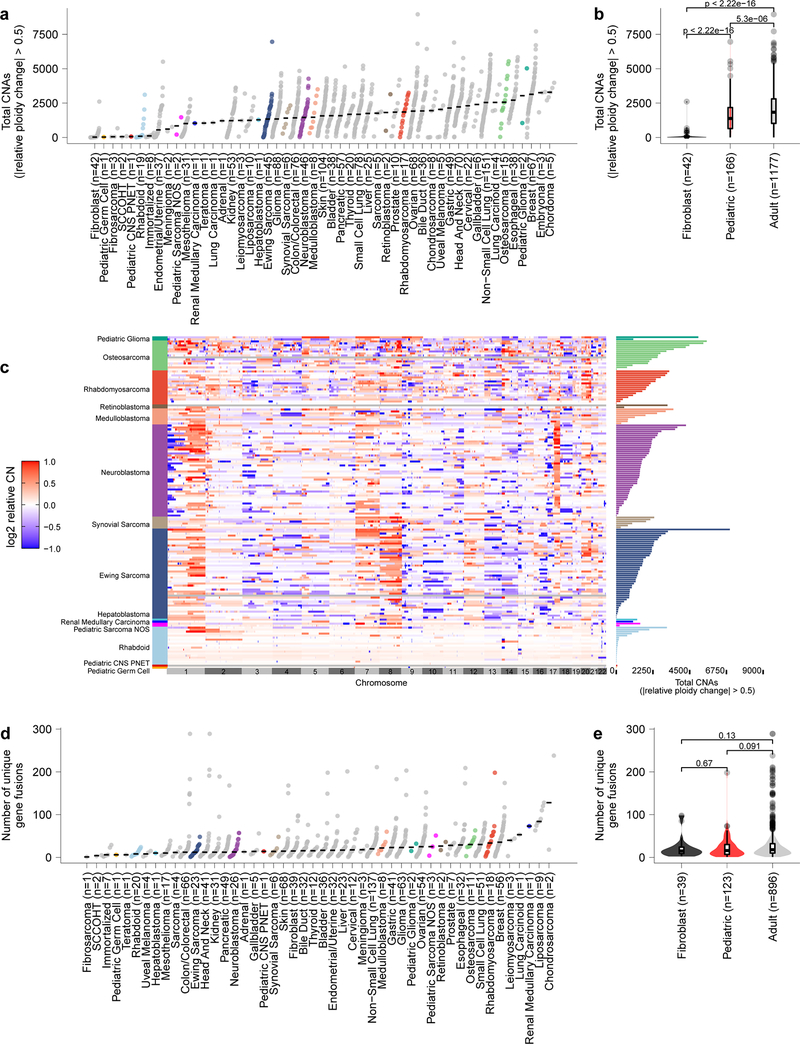
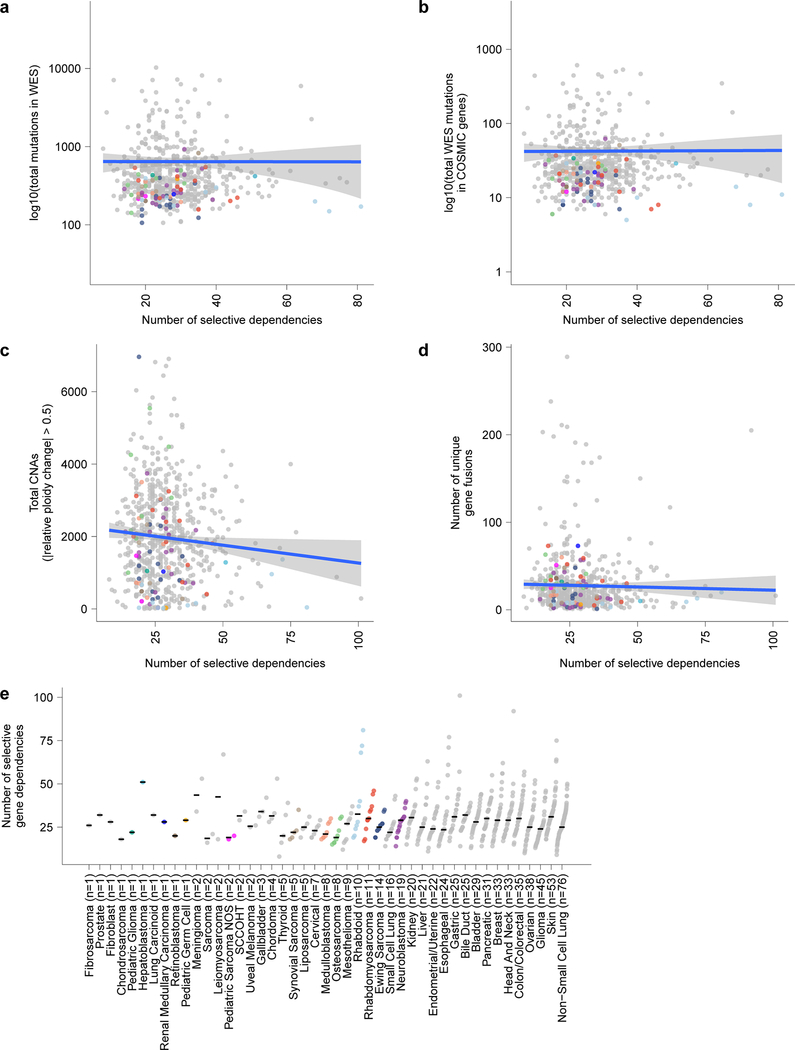
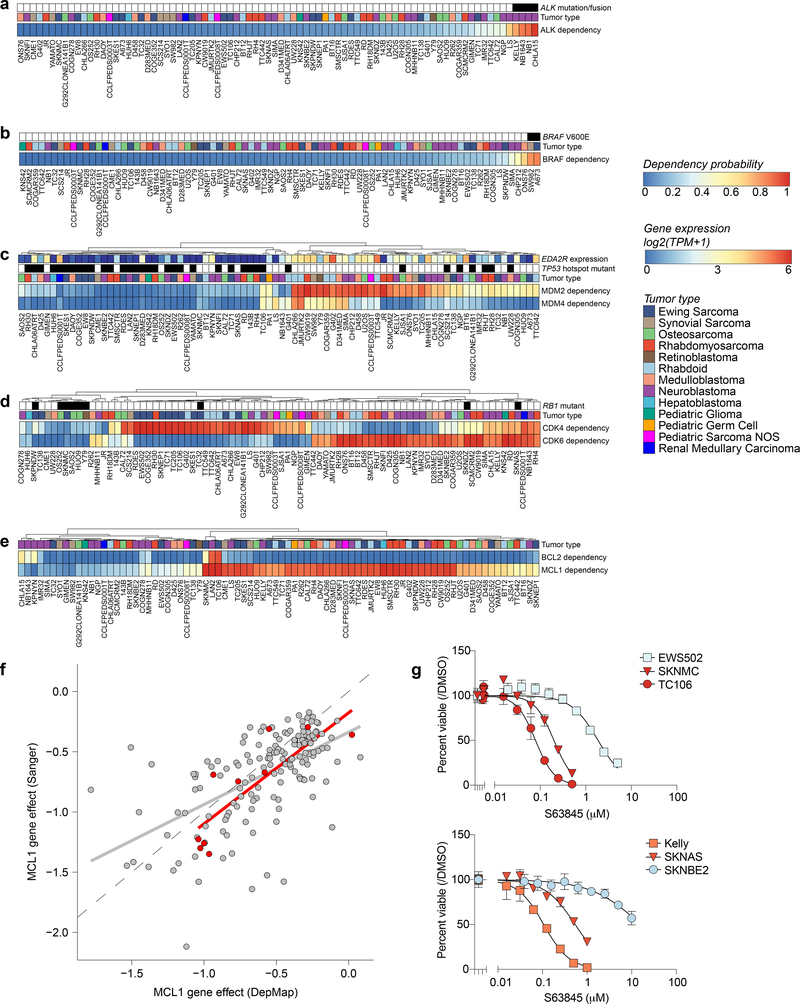
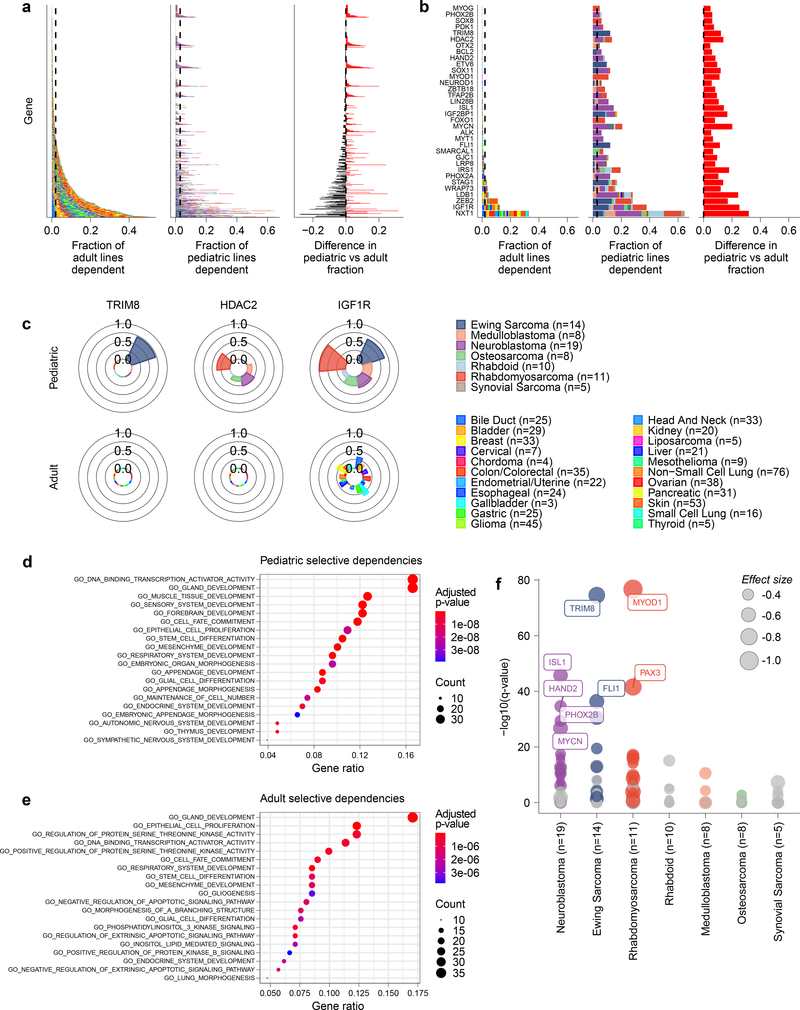
Exciting therapeutic targets are emerging from CRISPR-based screens of high mutational-burden adult cancers. A key question, however, is whether functional genomic approaches will yield new targets in pediatric cancers, known for remarkably few mutations, which often encode proteins considered challenging drug targets. To address this, we created a first-generation pediatric cancer dependency map representing 13 pediatric solid and brain tumor types. Eighty-two pediatric cancer cell lines were subjected to genome-scale CRISPR-Cas9 loss-of-function screening to identify genes required for cell survival. In contrast to the finding that pediatric cancers harbor fewer somatic mutations, we found a similar complexity of genetic dependencies in pediatric cancer cell lines compared to that in adult models. Findings from the pediatric cancer dependency map provide preclinical support for ongoing precision medicine clinical trials. The vulnerabilities observed in pediatric cancers were often distinct from those in adult cancer, indicating that repurposing adult oncology drugs will be insufficient to address childhood cancers.
Figures
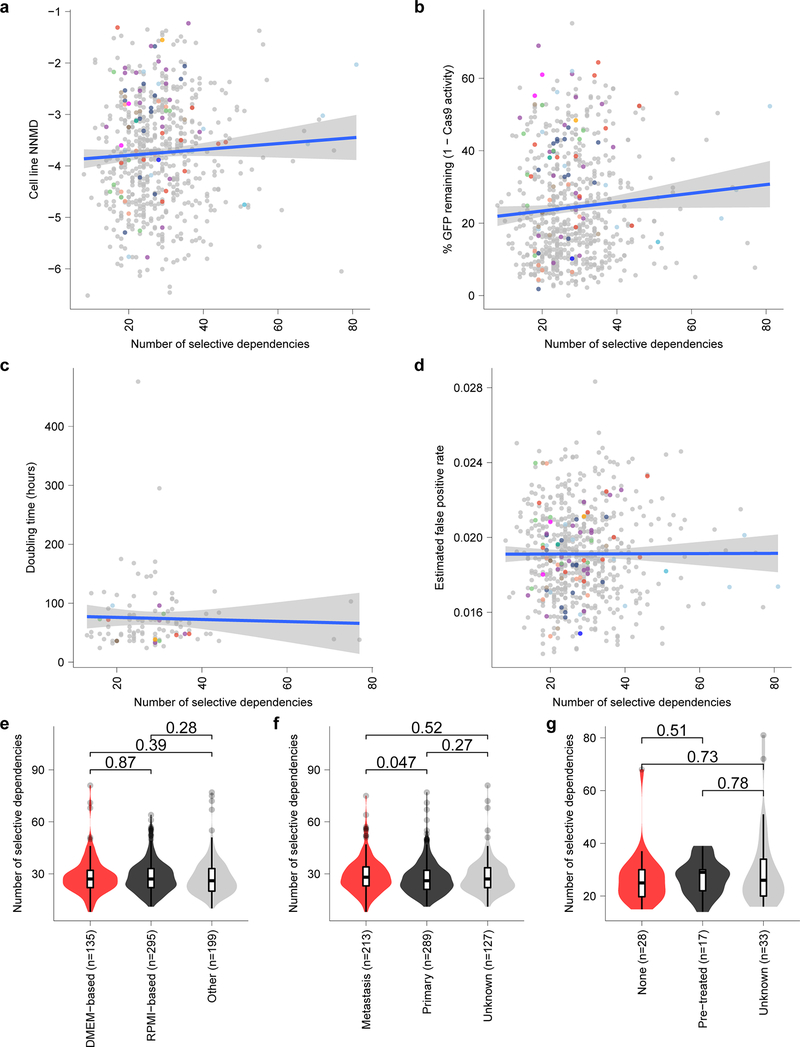
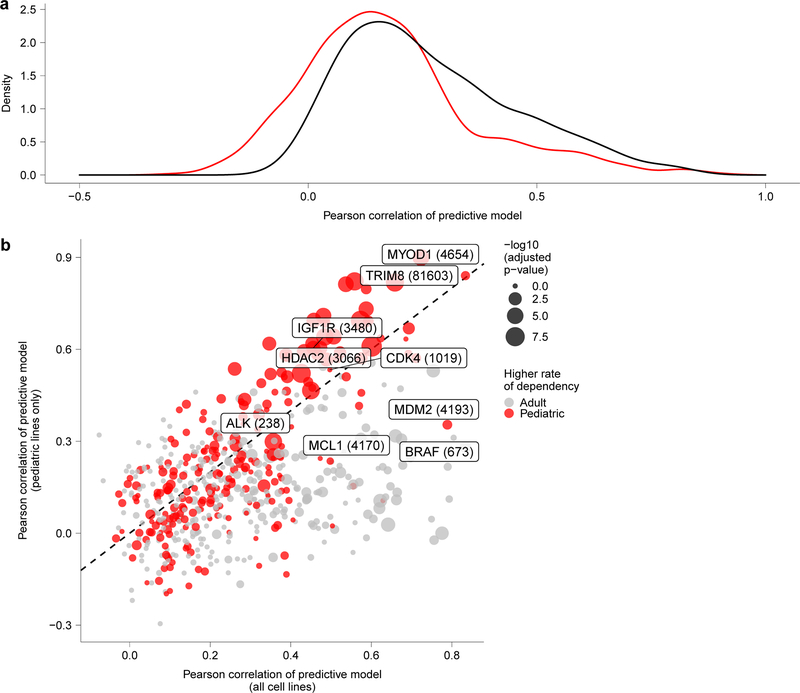
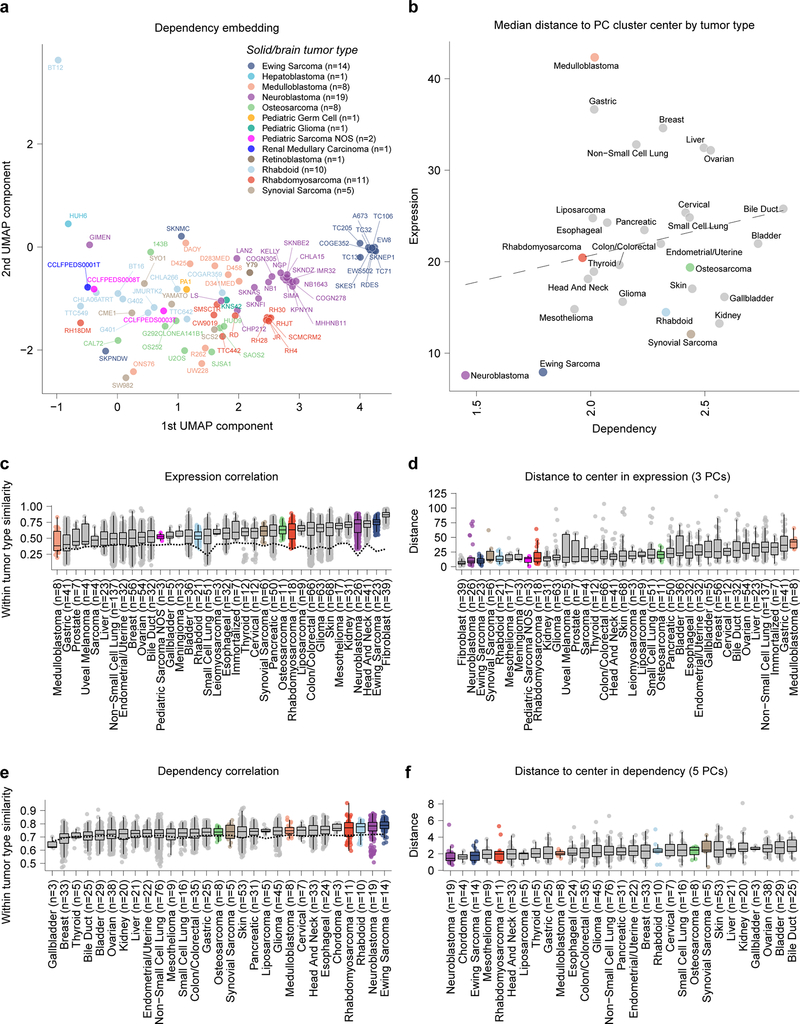
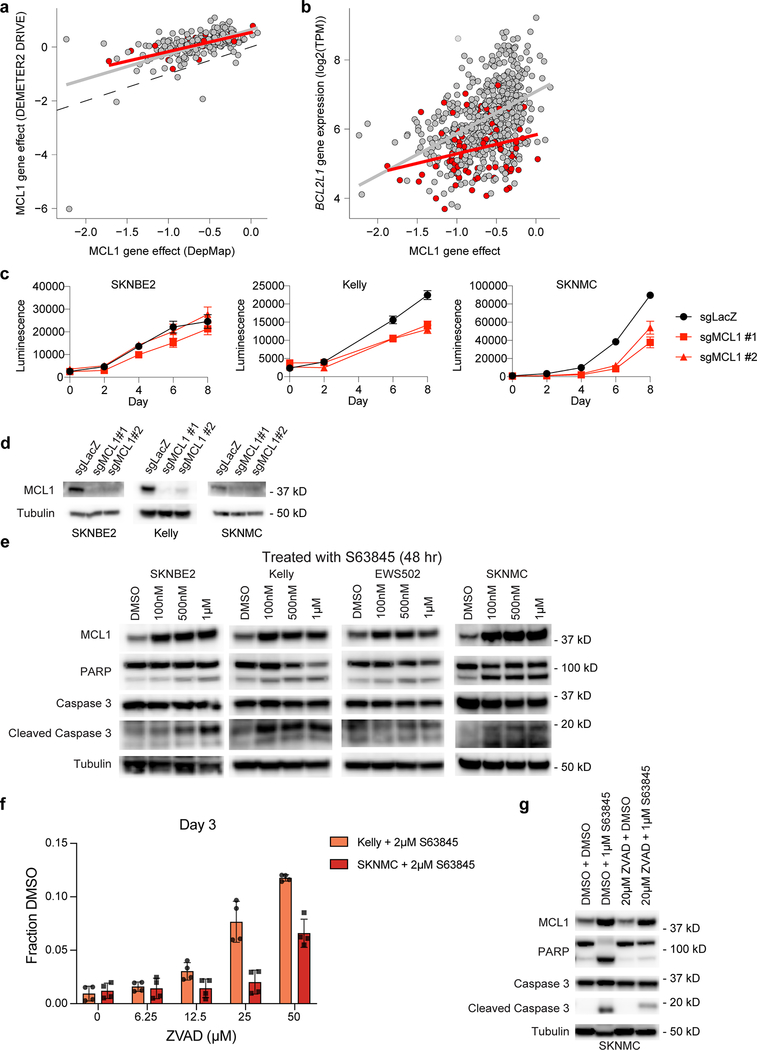
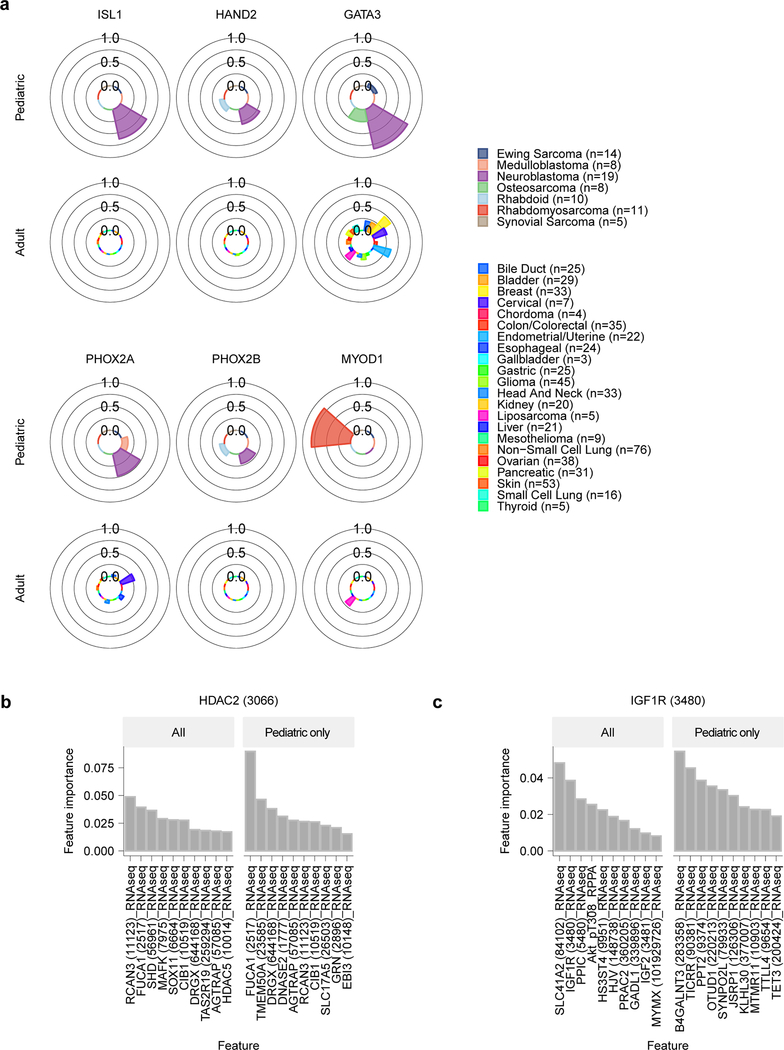
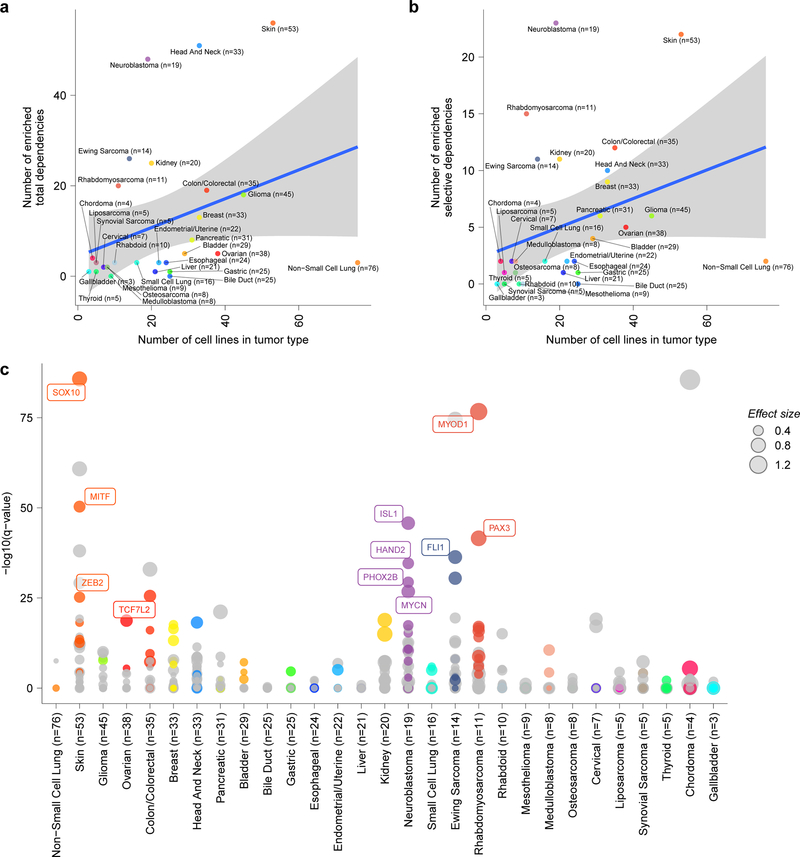
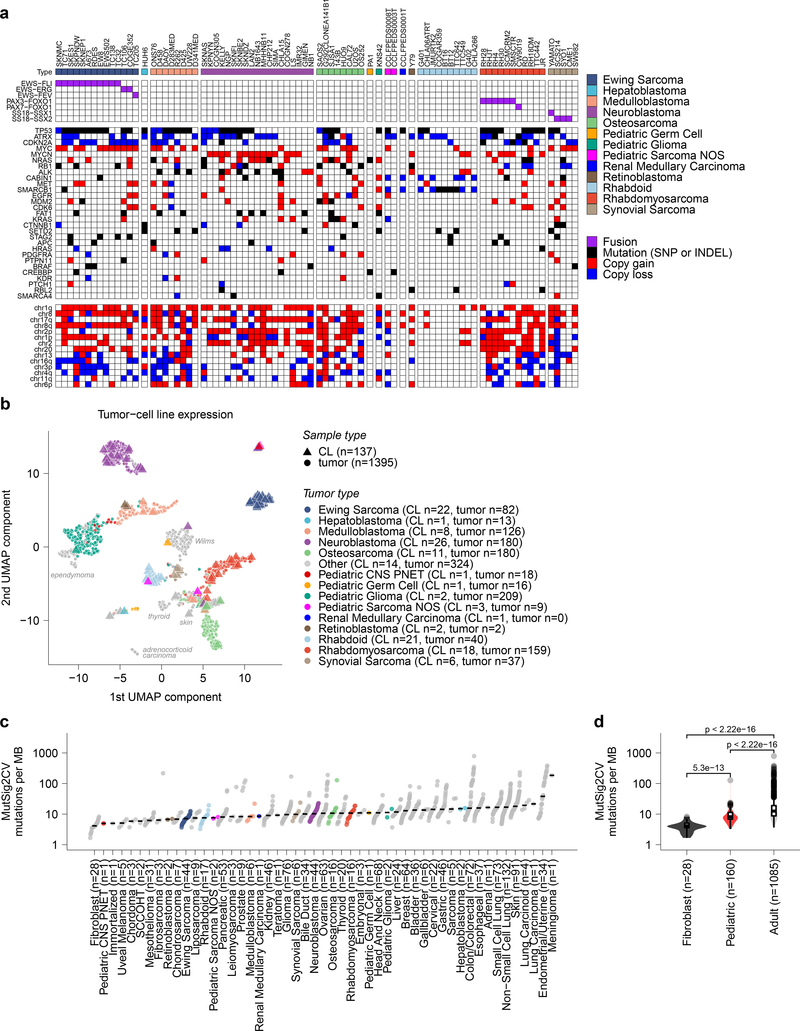
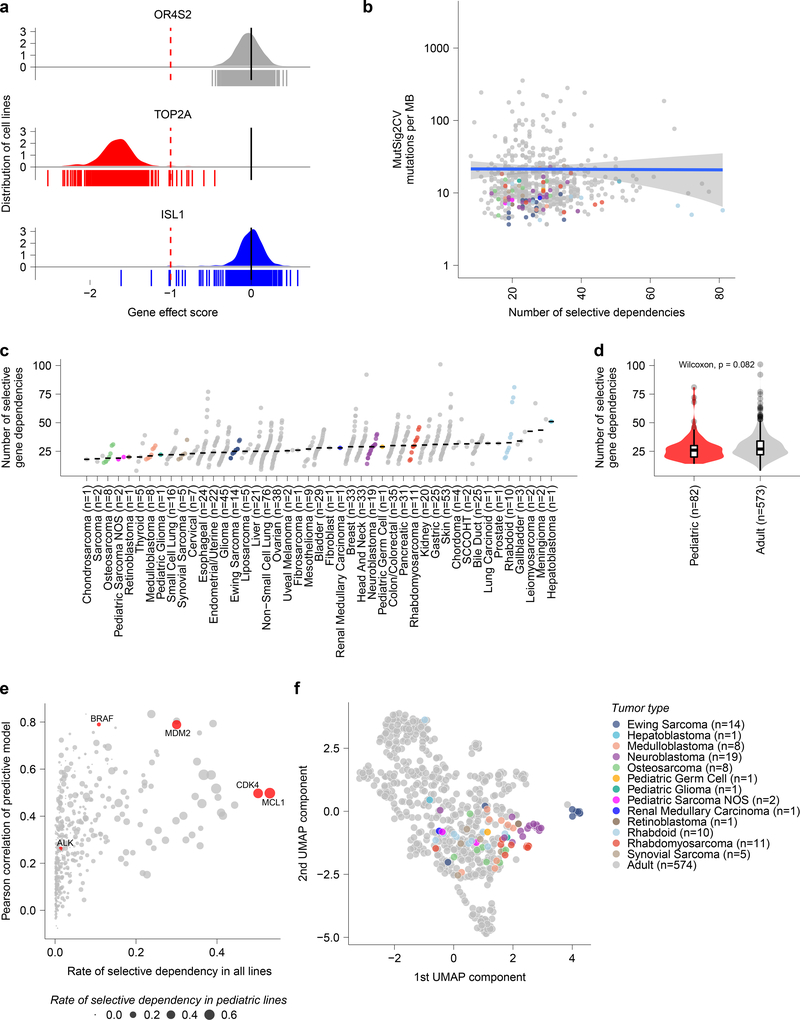
References
-
- Park JR et al. A phase III randomized clinical trial (RCT) of tandem myeloablative autologous stem cell transplant (ASCT) using peripheral blood stem cell (PBSC) as consolidation therapy for high-risk neuroblastoma (HR-NB): A Children’s Oncology Group (COG) study. JCO 34, LBA3–LBA3 (2016).
-
- Weigel BJ et al. Intensive Multiagent Therapy, Including Dose-Compressed Cycles of Ifosfamide/Etoposide and Vincristine/Doxorubicin/Cyclophosphamide, Irinotecan, and Radiation, in Patients With High-Risk Rhabdomyosarcoma: A Report From the Children’s Oncology Group. J. Clin. Oncol 34, 117–122 (2016). - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
