

Deficiency of telomere-associated repressor activator protein 1 precipitates cardiac aging in mice via p53/PPARα signaling
- PMID: 33754023
- PMCID: PMC7978321
- DOI: 10.7150/thno.51739
Deficiency of telomere-associated repressor activator protein 1 precipitates cardiac aging in mice via p53/PPARα signaling
Abstract
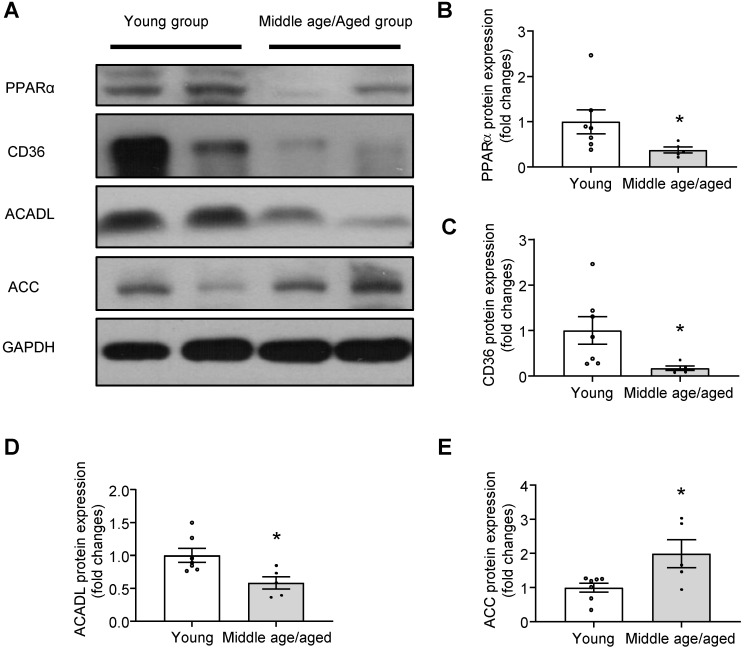
Background: Telomere shortening and dysfunction may cause metabolic disorders, tissue damage and age-dependent pathologies. However, little is known about the association of telomere-associated protein Rap1 with mitochondrial energy metabolism and cardiac aging. Methods: Echocardiography was performed to detect cardiac structure and function in Rap1+/+ and Rap1-/- mice at different ages (3 months, 12 months and 20 months). Telomere length, DNA damage, cardiac senescence and cardiomyocyte size were analyzed using the real-time PCR, Western blotting, senescence associated β-galactosidase assay and wheat germ agglutinin staining, respectively. Western blotting was also used to determine the level of cardiac fatty acid metabolism related key enzymes in mouse and human myocardium. Chromatin immunoprecipitation assay was used to verify the direct link between p53 and PPARα. The p53 inhibitor, Pifithrin-α and PPARα activator WY14643 were utilized to identify the effects of Rap1/p53/PPARα signaling pathway. Results: Telomere was shortened concomitant with extensive DNA damage in aged Rap1-/- mouse hearts, evidenced by reduced T/S ratios and increased nuclear γH2AX. Meanwhile, the aging-associated phenotypes were pronounced as reflected by altered mitochondrial ultrastructure, enhanced senescence, cardiac hypertrophy and dysfunction. Mechanistically, acetylated p53 and nuclear p53 was enhanced in the Rap1-/- mouse hearts, concomitant with reduced PPARα. Importantly, p53 directly binds to the promoter of PPARα in mouse hearts and suppresses the transcription of PPARα. In addition, aged Rap1-/- mice exhibited reduced cardiac fatty acid metabolism. Pifithrin-α alleviated cardiac aging and enhanced fatty acid metabolism in the aged Rap1-/- mice. Activating PPARα with WY14643 in primarily cultured Rap1-/- cardiomyocytes restored maximal oxygen consumption rates. Reduced Rap1 expression and impaired p53/PPARα signaling also presented in aged human myocardium. Conclusion: In summary, Rap1 may link telomere biology to fatty acid metabolism and aging-related cardiac pathologies via modulating the p53/PPARα signaling pathway, which could represent a therapeutic target in preventing/attenuating cardiac aging.
Keywords: PPARα; Rap1; cardiac aging; fatty acid metabolism; p53.
© The author(s).
Conflict of interest statement
Competing Interests: The authors have declared that no competing interest exists.
Figures






References
-
- Wagner JUG, Dimmeler S. Cellular cross-talks in the diseased and aging heart. J Mol Cell Cardiol. 2020;138:136–46. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
