

A novel role of kallikrein-related peptidase 8 in the pathogenesis of diabetic cardiac fibrosis
- PMID: 33754057
- PMCID: PMC7977470
- DOI: 10.7150/thno.48530
A novel role of kallikrein-related peptidase 8 in the pathogenesis of diabetic cardiac fibrosis
Abstract
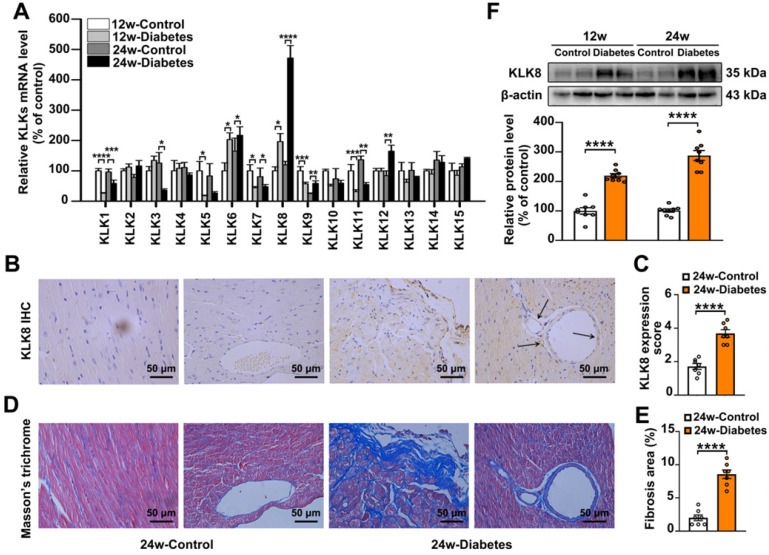
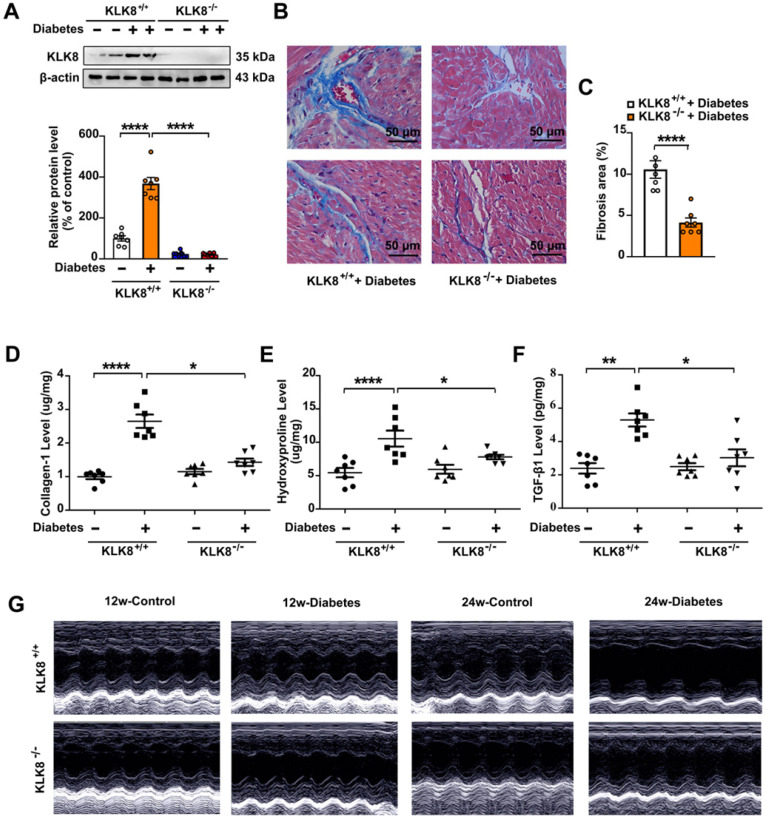
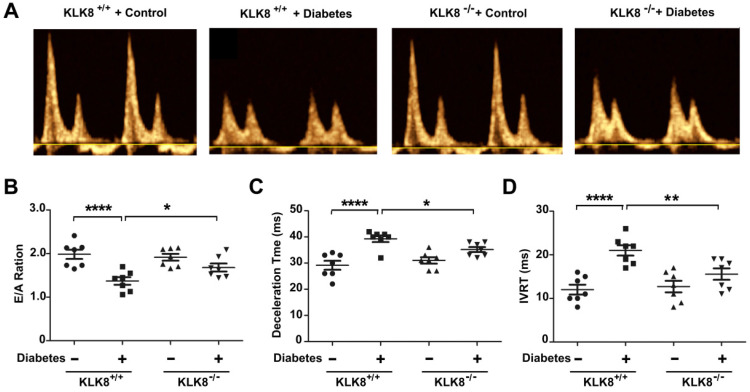
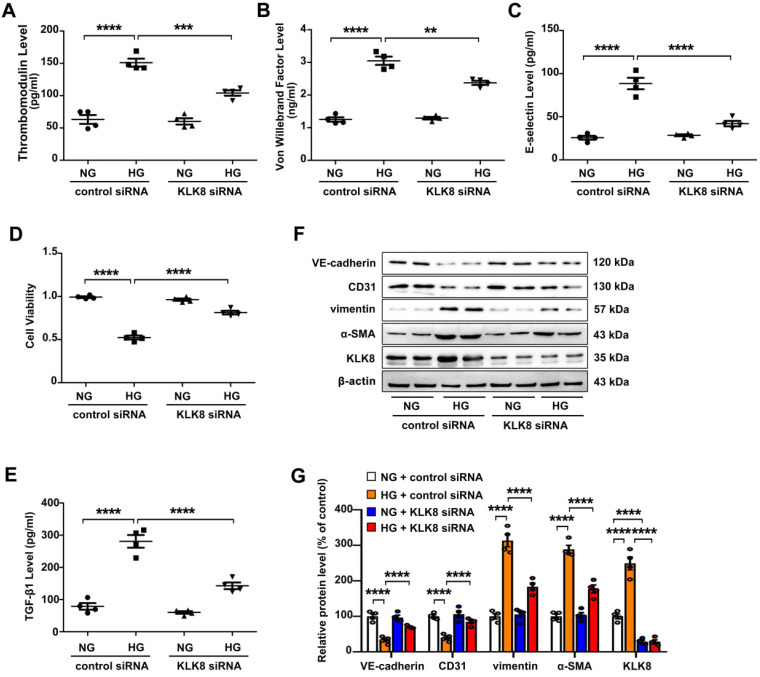
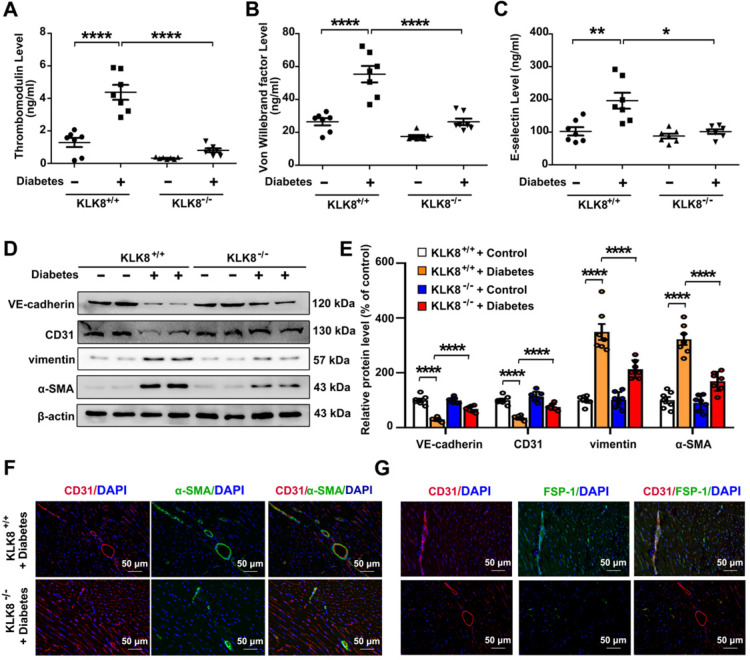
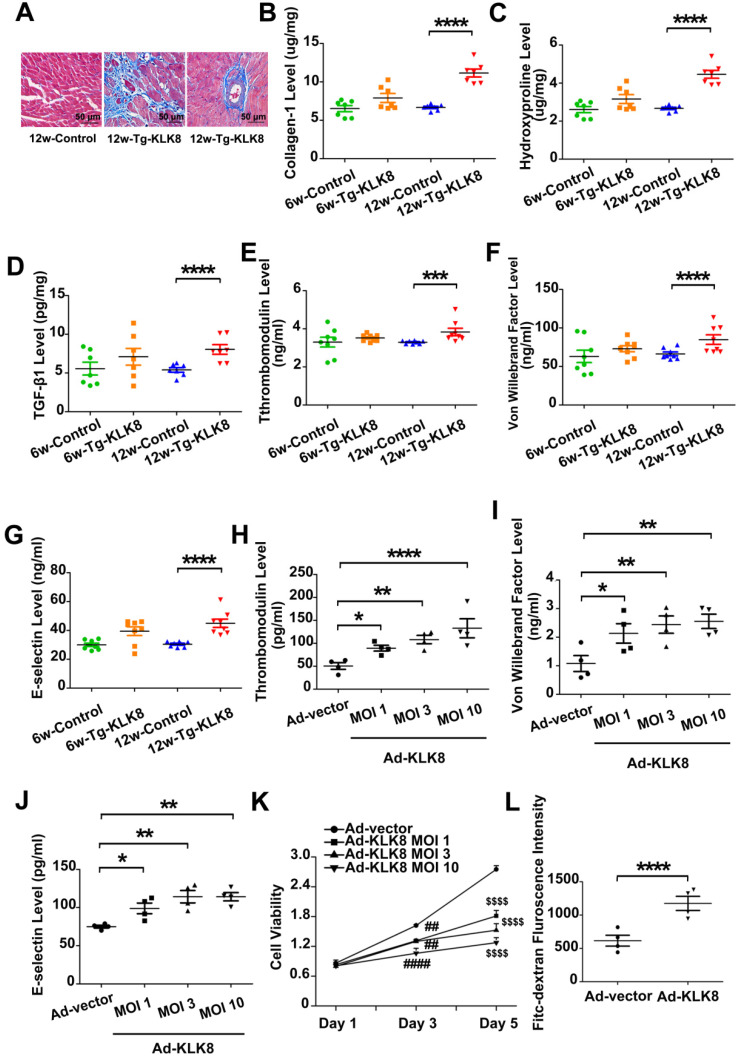
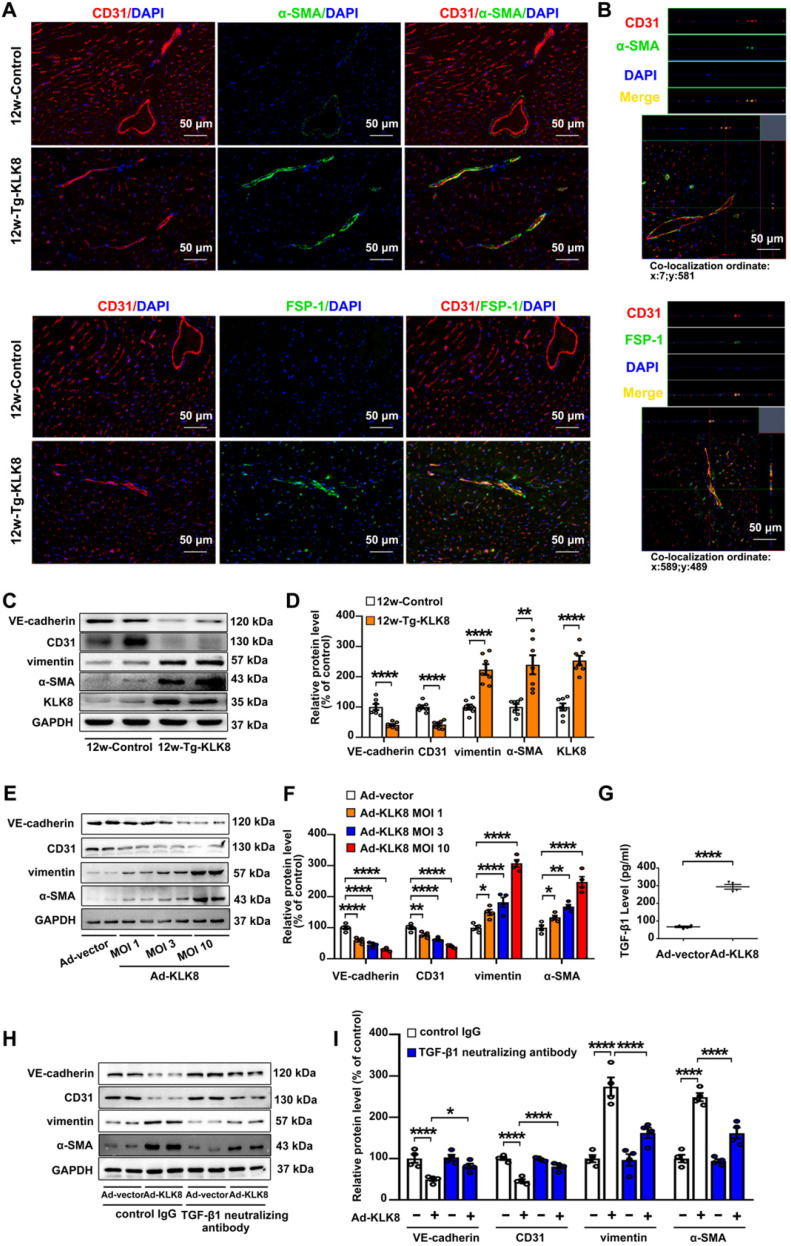
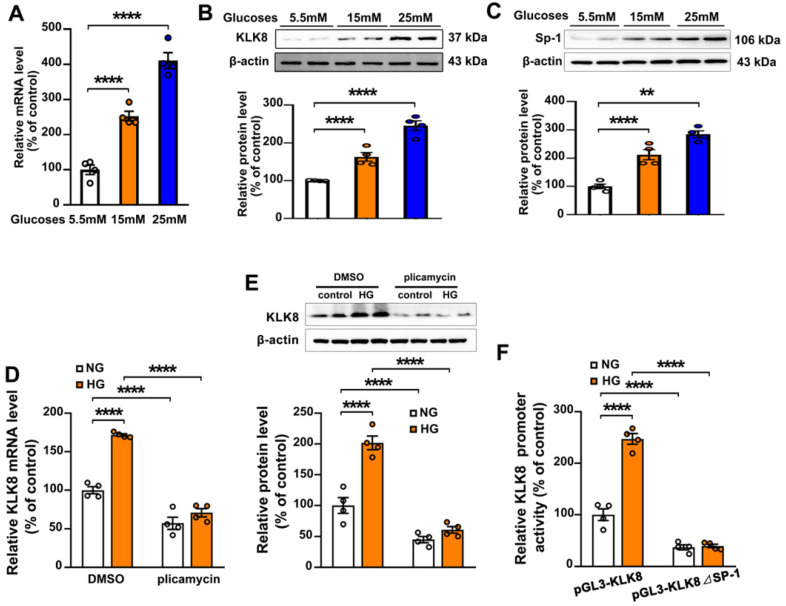
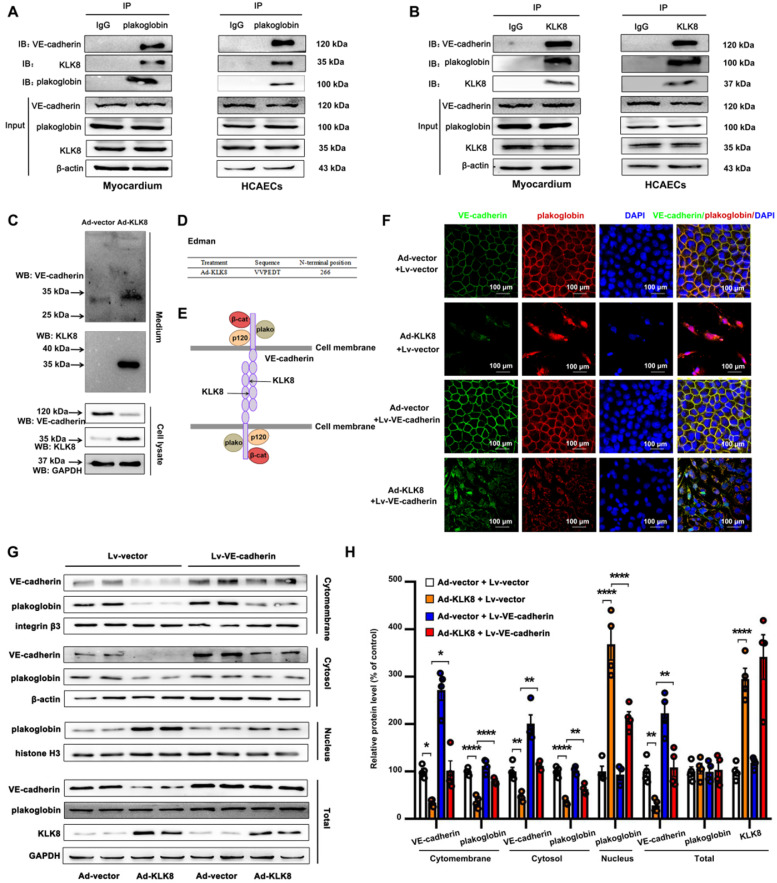
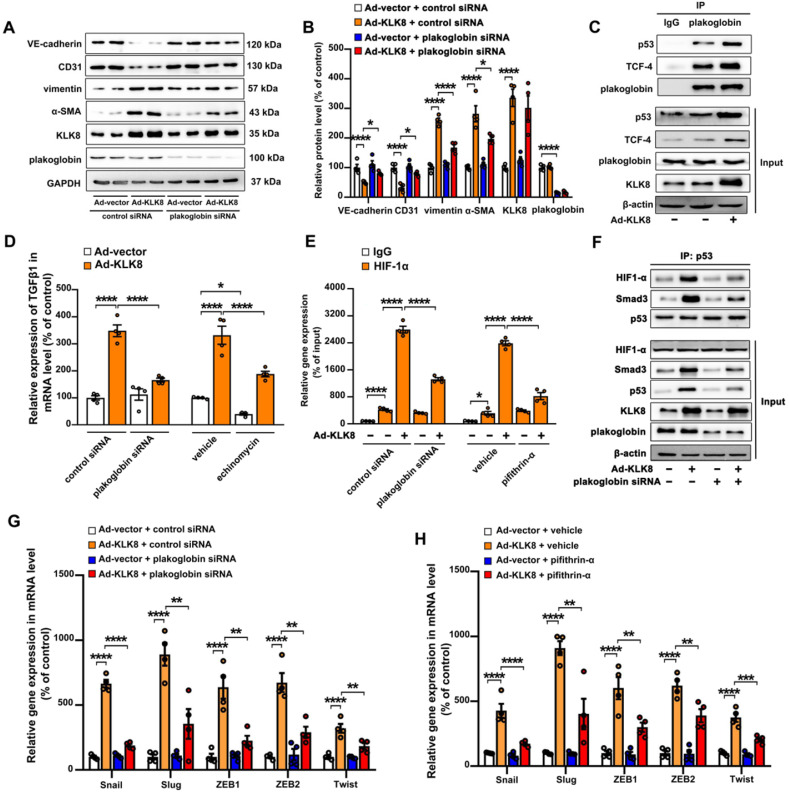
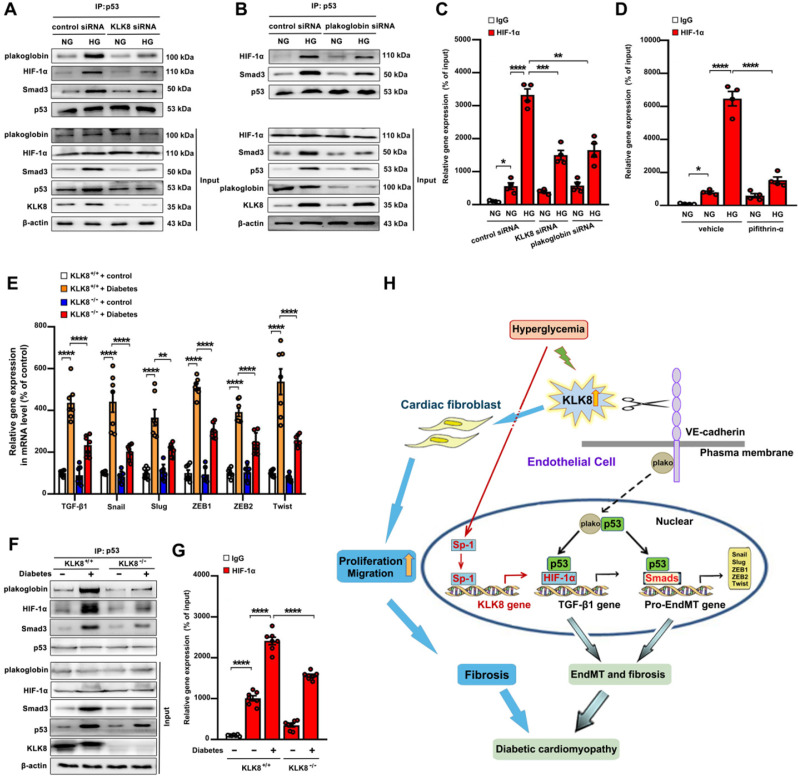
Rationale: Among all the diabetic complications, diabetic cardiomyopathy, which is characterized by myocyte loss and myocardial fibrosis, is the leading cause of mortality and morbidity in diabetic patients. Tissue kallikrein-related peptidases (KLKs) are secreted serine proteases, that have distinct and overlapping roles in the pathogenesis of cardiovascular diseases. However, whether KLKs are involved in the development of diabetic cardiomyopathy remains unknown.The present study aimed to determine the role of a specific KLK in the initiation of endothelial-to-mesenchymal transition (EndMT) during the pathogenesis of diabetic cardiomyopathy. Methods and Results-By screening gene expression profiles of KLKs, it was found that KLK8 was highly induced in the myocardium of mice with streptozotocin-induced diabetes. KLK8 deficiency attenuated diabetic cardiac fibrosis, and rescued the impaired cardiac function in diabetic mice. Small interfering RNA (siRNA)-mediated KLK8 knockdown significantly attenuated high glucose-induced endothelial damage and EndMT in human coronary artery endothelial cells (HCAECs). Diabetes-induced endothelial injury and cardiac EndMT were significantly alleviated in KLK8-deficient mice. In addition, transgenic overexpression of KLK8 led to interstitial and perivascular cardiac fibrosis, endothelial injury and EndMT in the heart. Adenovirus-mediated overexpression of KLK8 (Ad-KLK8) resulted in increases in endothelial cell damage, permeability and transforming growth factor (TGF)-β1 release in HCAECs. KLK8 overexpression also induced EndMT in HCAECs, which was alleviated by a TGF-β1-neutralizing antibody. A specificity protein-1 (Sp-1) consensus site was identified in the human KLK8 promoter and was found to mediate the high glucose-induced KLK8 expression. Mechanistically, it was identified that the vascular endothelial (VE)-cadherin/plakoglobin complex may associate with KLK8 in HCAECs. KLK8 cleaved the VE-cadherin extracellular domain, thus promoting plakoglobin nuclear translocation. Plakoglobin was required for KLK8-induced EndMT by cooperating with p53. KLK8 overexpression led to plakoglobin-dependent association of p53 with hypoxia inducible factor (HIF)-1α, which further enhanced the transactivation effect of HIF-1α on the TGF-β1 promoter. KLK8 also induced the binding of p53 with Smad3, subsequently promoting pro-EndMT reprogramming via the TGF-β1/Smad signaling pathway in HCAECs. The in vitro and in vivo findings further demonstrated that high glucose may promote plakoglobin-dependent cooperation of p53 with HIF-1α and Smad3, subsequently increasing the expression of TGF-β1 and the pro-EndMT target genes of the TGF-β1/Smad signaling pathway in a KLK8-dependent manner. Conclusions: The present findings uncovered a novel pro-EndMT mechanism during the pathogenesis of diabetic cardiac fibrosis via the upregulation of KLK8, and may contribute to the development of future KLK8-based therapeutic strategies for diabetic cardiomyopathy.
Keywords: KLK8; cardiac fibrosis; diabetic cardiomyopathy; endothelial-to-mesenchymal transition; plakoglobin.
© The author(s).
Conflict of interest statement
Competing Interests: The authors have declared that no competing interest exists.
Figures
References
-
- Zheng Y, Ley SH, Hu FB. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat Rev Endocrinol. 2018;14:88–98. - PubMed
-
- Prattichizzo F, Matacchione G, Giuliani A, Sabbatinelli J, Olivieri F, de Candia P. et al. Extracellular vesicle-shuttled miRNAs: a critical appraisal of their potential as nano-diagnostics and nano-therapeutics in type 2 diabetes mellitus and its cardiovascular complications. Theranostics. 2021;11:1031–45. - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
