

The role of GLS1-mediated glutaminolysis/2-HG/H3K4me3 and GSH/ROS signals in Th17 responses counteracted by PPARγ agonists
- PMID: 33754076
- PMCID: PMC7977454
- DOI: 10.7150/thno.54803
The role of GLS1-mediated glutaminolysis/2-HG/H3K4me3 and GSH/ROS signals in Th17 responses counteracted by PPARγ agonists
Abstract
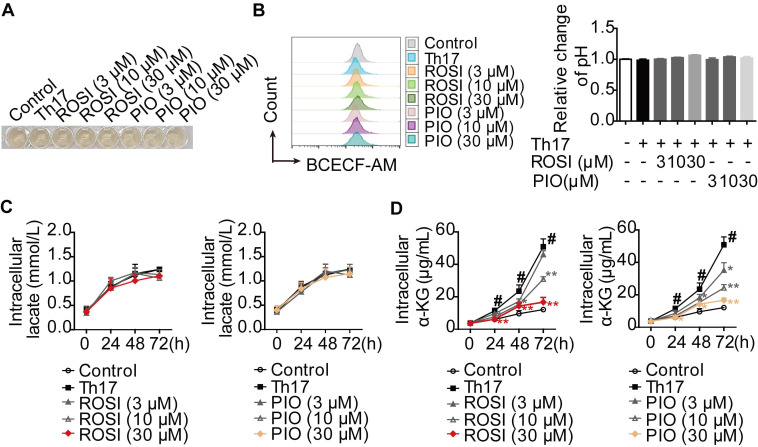
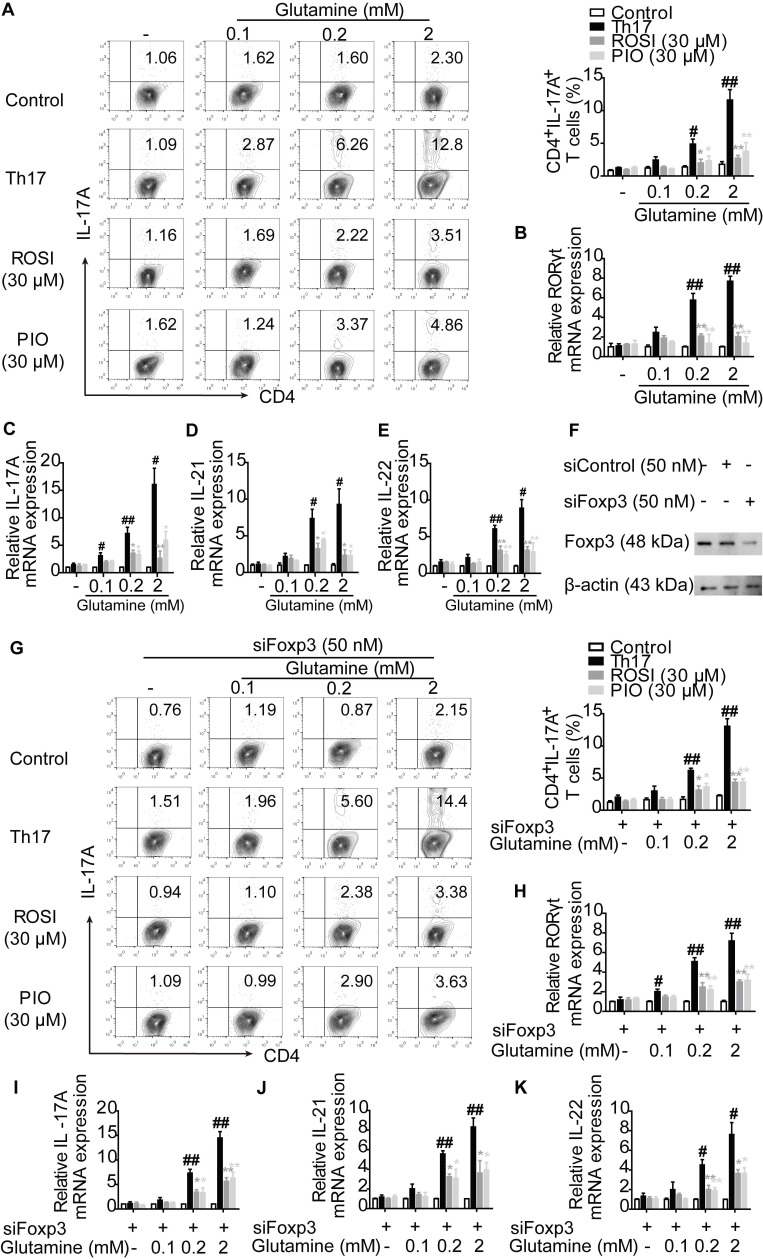
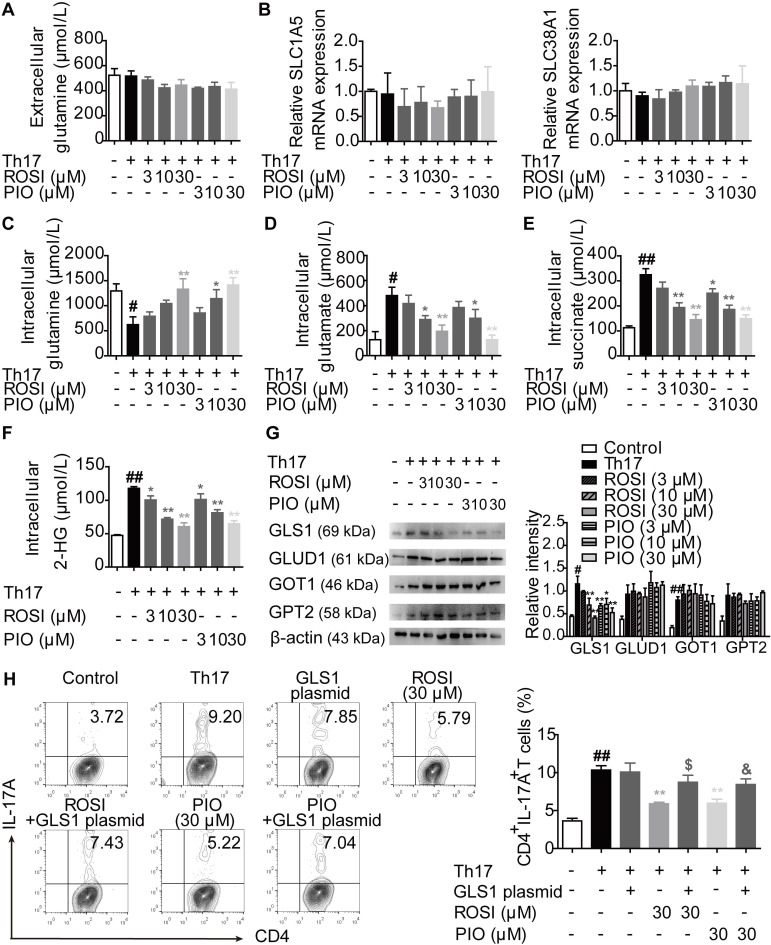
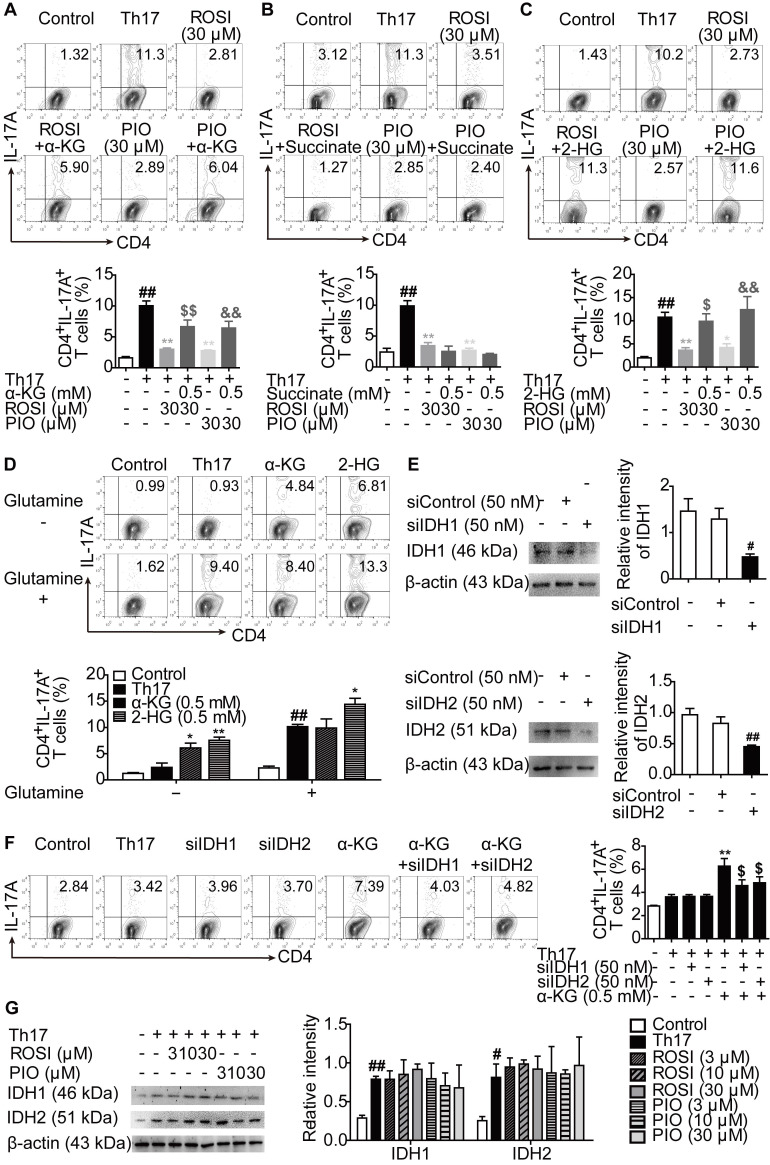
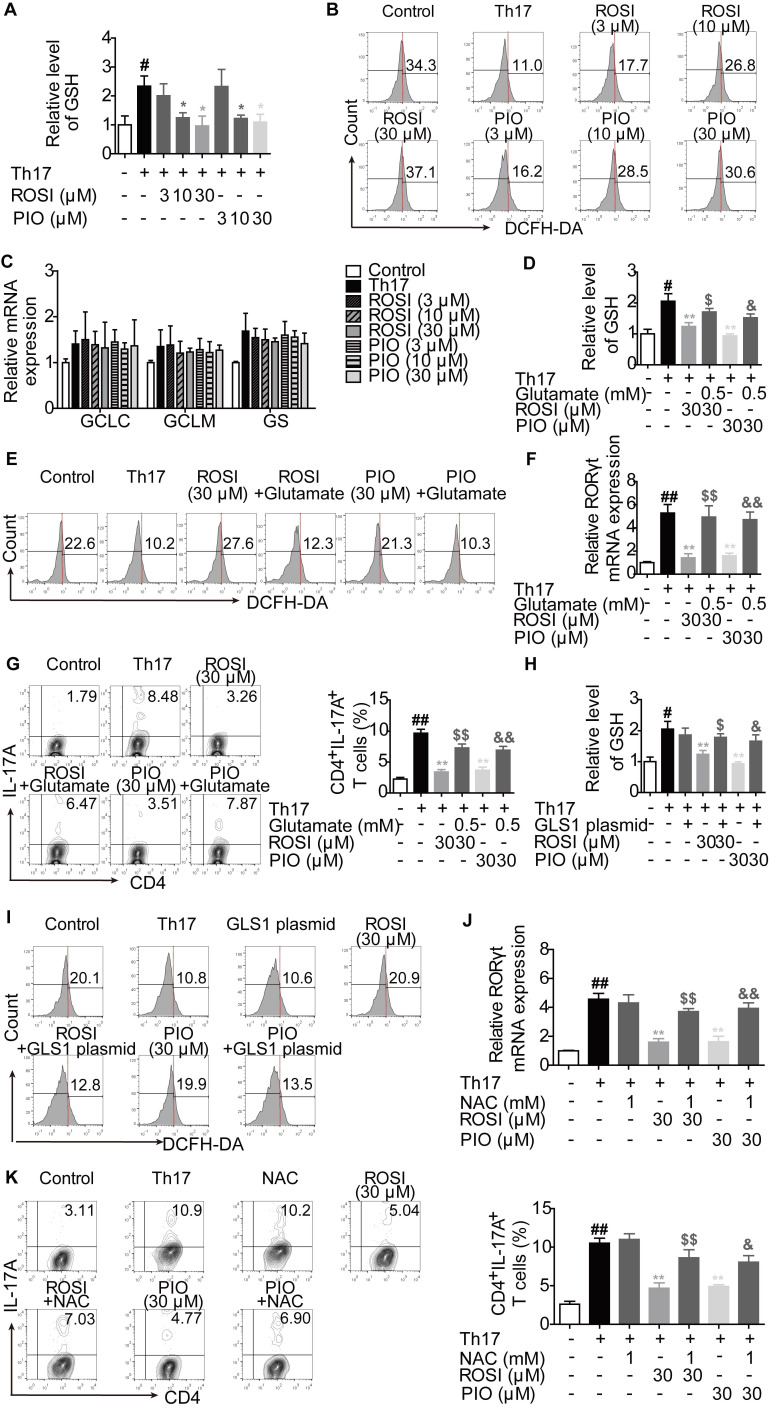
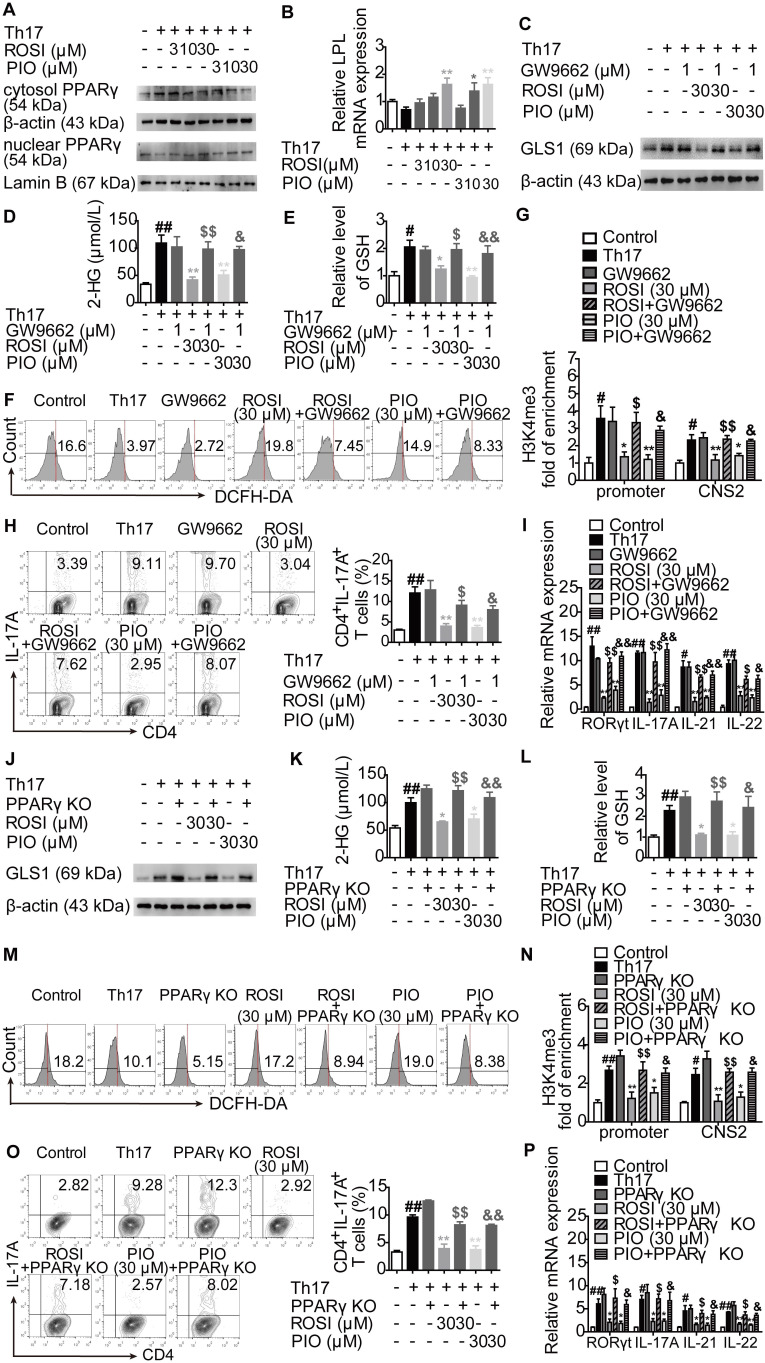
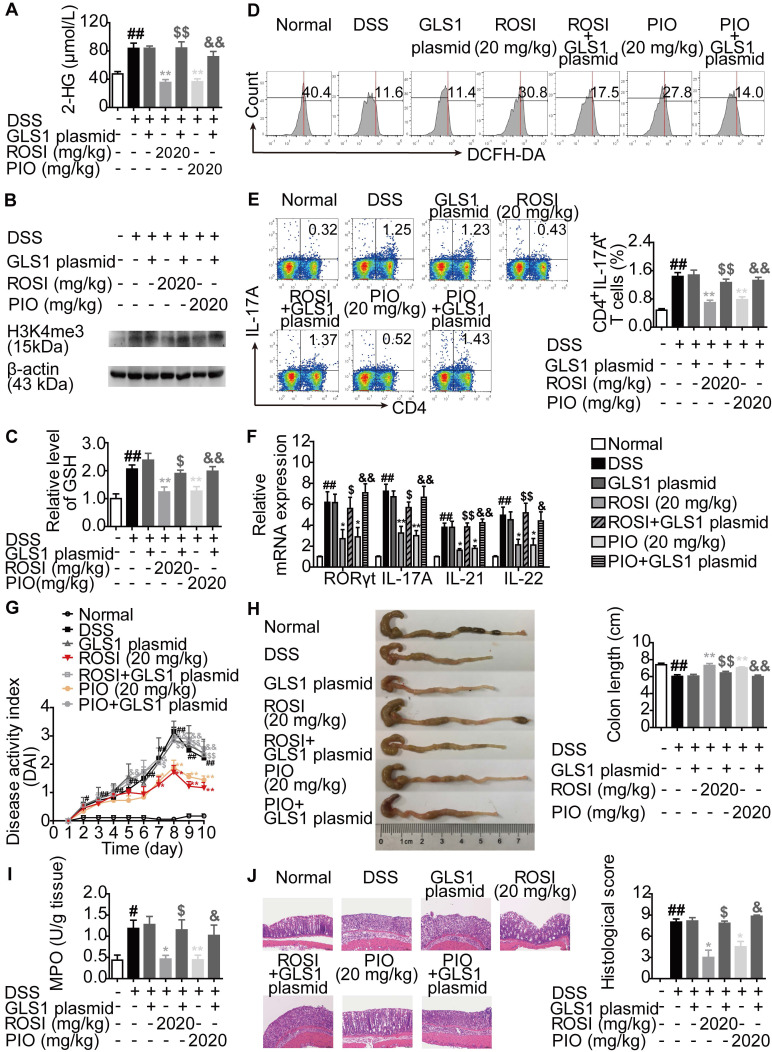
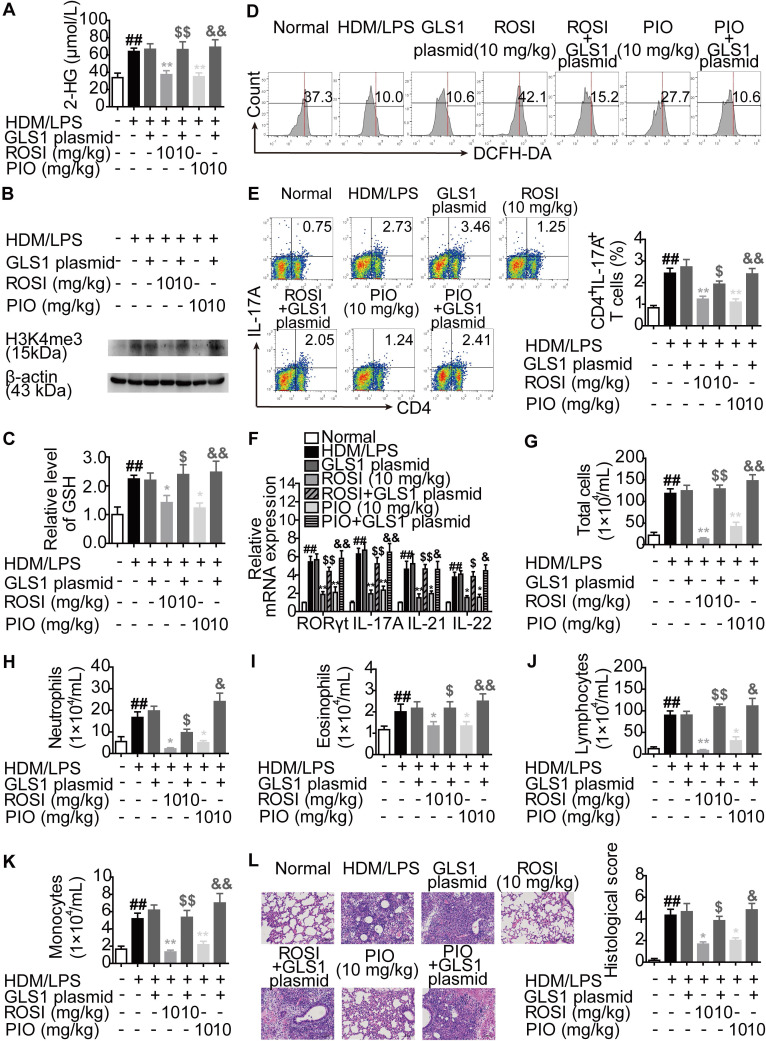
Background: Peroxisome proliferator-activated receptor gamma (PPARγ) has the ability to counter Th17 responses, but the full mechanisms remain elusive. Herein, we aimed to elucidate this process in view of cellular metabolism, especially glutaminolysis. Methods: MTT, CCK-8, Annexin V-FITC/PI staining or trypan blue exclusion assays were used to analyze cytotoxicity. Flow cytometry and Q-PCR assays were applied to determine Th17 responses. The detection of metabolite levels using commercial kits and rate-limiting enzyme expression using western blotting assays was performed to illustrate the metabolic activity. ChIP assays were used to examine H3K4me3 modifications. Mouse models of dextran sulfate sodium (DSS)-induced colitis and house dust mite (HDM)/lipopolysaccharide (LPS)-induced asthma were established to confirm the mechanisms studied in vitro. Results: The PPARγ agonists rosiglitazone and pioglitazone blocked glutaminolysis but not glycolysis under Th17-skewing conditions, as indicated by the detection of intracellular lactate and α-KG and the fluorescence ratios of BCECF-AM. The PPARγ agonists prevented the utilization of glutamine and thus directly limited Th17 responses even when Foxp3 was deficient. The mechanisms were ascribed to restricted conversion of glutamine to glutamate by reducing the expression of the rate-limiting enzyme GLS1, which was confirmed by GLS1 overexpression. Replenishment of α-KG and 2-HG but not succinate weakened the effects of PPARγ agonists, and α-KG-promoted Th17 responses were dampened by siIDH1/2. Inhibition of KDM5 but not KDM4/6 restrained the inhibitory effect of PPARγ agonists on IL-17A expression, and the H3K4me3 level in the promoter and CNS2 region of the il-17 gene locus down-regulated by PPARγ agonists was rescued by 2-HG and GLS1 overexpression. However, the limitation of PPARγ agonists on the mRNA expression of RORγt was unable to be stopped by 2-HG but was attributed to GSH/ROS signals subsequent to GLS1. The exact role of PPARγ was proved by GW9662 or PPARγ knockout, and the mechanisms for PPARγ-inhibited Th17 responses were further confirmed by GLS1 overexpression in vivo. Conclusion: PPARγ agonists repressed Th17 responses by counteracting GLS1-mediated glutaminolysis/2-HG/H3K4me3 and GSH/ROS signals, which is beneficial for Th17 cell-related immune dysregulation.
Keywords: PPARγ; Th17 responses; glutaminase 1; glutaminolysis.
© The author(s).
Conflict of interest statement
Competing Interests: The authors have declared that no competing interest exists.
Figures

References
-
- Jiang Y, Wang X, Dong C. Molecular mechanisms of T helper 17 cell differentiation: Emerging roles for transcription cofactors. Adv Immunol. 2019;144:121–153. - PubMed
-
- de Oliveira Boldrini V, Dos Santos Farias A, Degasperi GR. Deciphering targets of Th17 cells fate: From metabolism to nuclear receptors. Scand J Immunol. 2019;90:e12793. - PubMed
-
- Leppkes M, Becker C, Ivanov II, Hirth S, Wirtz S, Neufert C. et al. RORgamma-expressing Th17 cells induce murine chronic intestinal inflammation via redundant effects of IL-17A and IL-17F. Gastroenterology. 2009;136:257–267. - PubMed
-
- Chenuet P, Fauconnier L, Madouri F, Marchiol T, Rouxel N, Ledru A. et al. Neutralization of either IL-17A or IL-17F is sufficient to inhibit house dust mite induced allergic asthma in mice. Clin Sci (Lond) 2017;131:2533–2548. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
