

Genomic Surveillance of a Globally Circulating Distinct Group W Clonal Complex 11 Meningococcal Variant, New Zealand, 2013-2018
- PMID: 33754994
- PMCID: PMC8007299
- DOI: 10.3201/eid2704.191716
Genomic Surveillance of a Globally Circulating Distinct Group W Clonal Complex 11 Meningococcal Variant, New Zealand, 2013-2018
Abstract
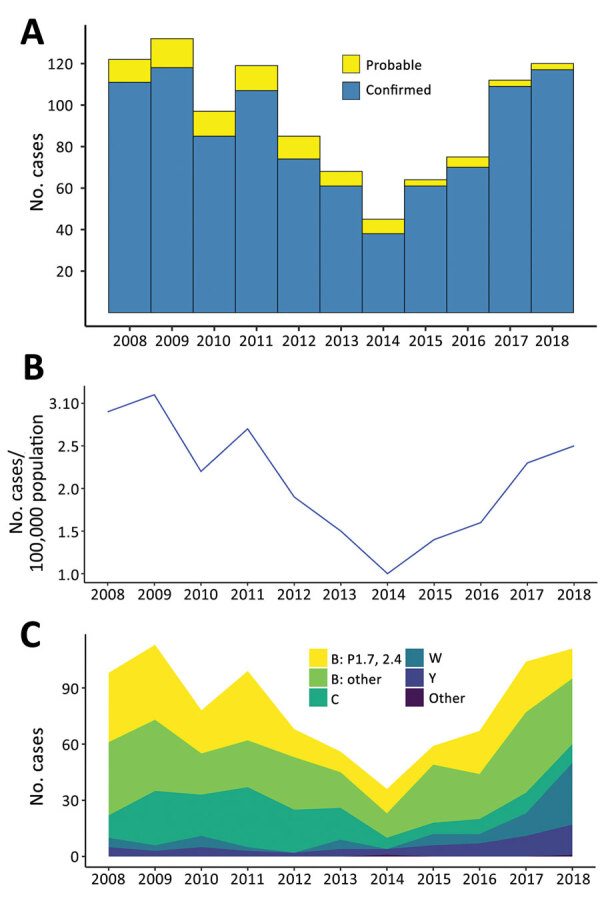
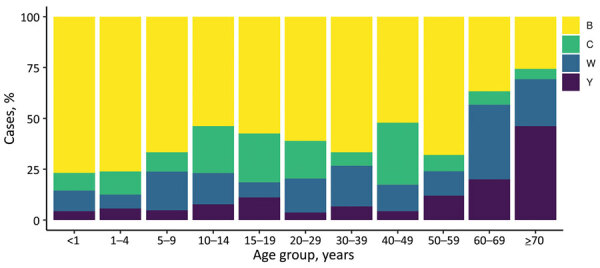
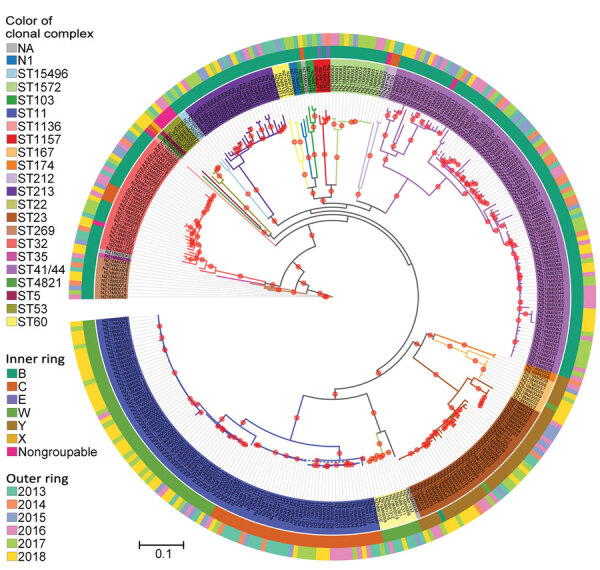
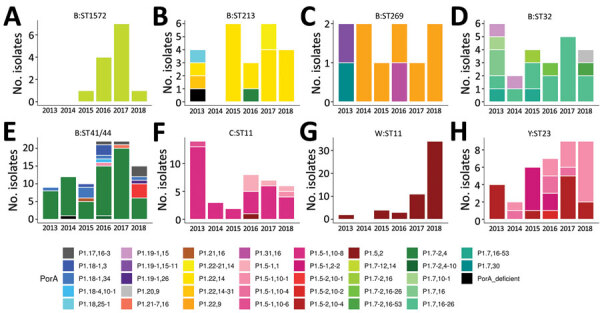
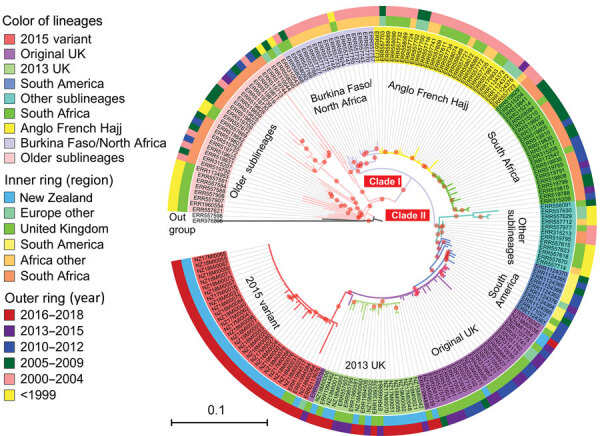
Genomic surveillance is an essential part of effective disease control, enabling identification of emerging and expanding strains and monitoring of subsequent interventions. Whole-genome sequencing was used to analyze the genomic diversity of all Neisseria meningitidis isolates submitted to the New Zealand Meningococcal Reference Laboratory during 2013-2018. Of the 347 isolates submitted for whole-genome sequencing, we identified 68 sequence types belonging to 18 clonal complexes (CC). The predominant CC was CC41/44; next in predominance was CC11. Comparison of the 45 New Zealand group W CC11 isolates with worldwide representatives of group W CC11 isolates revealed that the original UK strain, the 2013 UK strain, and a distinctive variant (the 2015 strain) were causing invasive group W meningococcal disease in New Zealand. The 2015 strain also demonstrated increased resistance to penicillin and has been circulating in Canada and several countries in Europe, highlighting that close monitoring is needed to prevent future outbreaks around the world.
Keywords: Neisseria meningitidis; New Zealand; Public Health Surveillance; bacteria; evolution; group W; invasive meningococcal disease; meningitis/encephalitis; meningococcal disease; whole-genome sequencing.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
