

Allosteric communication in class A β-lactamases occurs via cooperative coupling of loop dynamics
- PMID: 33755013
- PMCID: PMC8060031
- DOI: 10.7554/eLife.66567
Allosteric communication in class A β-lactamases occurs via cooperative coupling of loop dynamics
Abstract
Understanding allostery in enzymes and tools to identify it offer promising alternative strategies to inhibitor development. Through a combination of equilibrium and nonequilibrium molecular dynamics simulations, we identify allosteric effects and communication pathways in two prototypical class A β-lactamases, TEM-1 and KPC-2, which are important determinants of antibiotic resistance. The nonequilibrium simulations reveal pathways of communication operating over distances of 30 Å or more. Propagation of the signal occurs through cooperative coupling of loop dynamics. Notably, 50% or more of clinically relevant amino acid substitutions map onto the identified signal transduction pathways. This suggests that clinically important variation may affect, or be driven by, differences in allosteric behavior, providing a mechanism by which amino acid substitutions may affect the relationship between spectrum of activity, catalytic turnover, and potential allosteric behavior in this clinically important enzyme family. Simulations of the type presented here will help in identifying and analyzing such differences.
Keywords: KPC-2; TEM-1; medicine; molecular biophysics; structural biology; β-lactamase.
Plain language summary
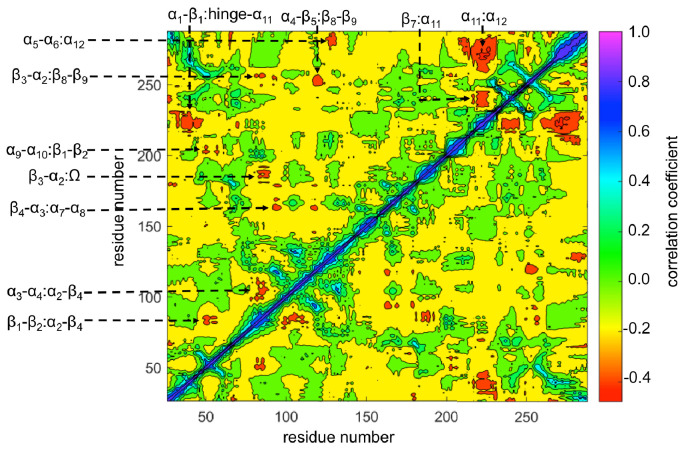
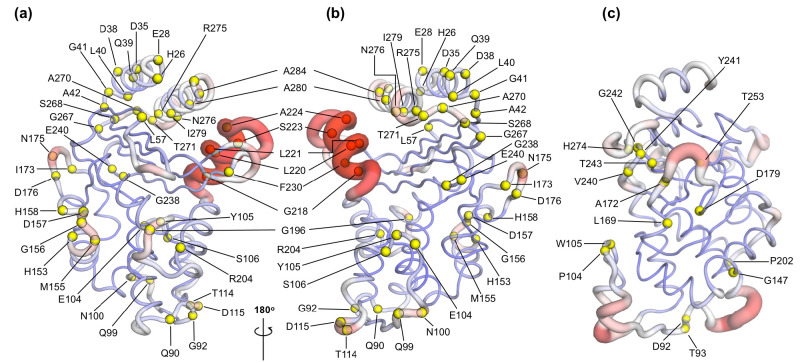
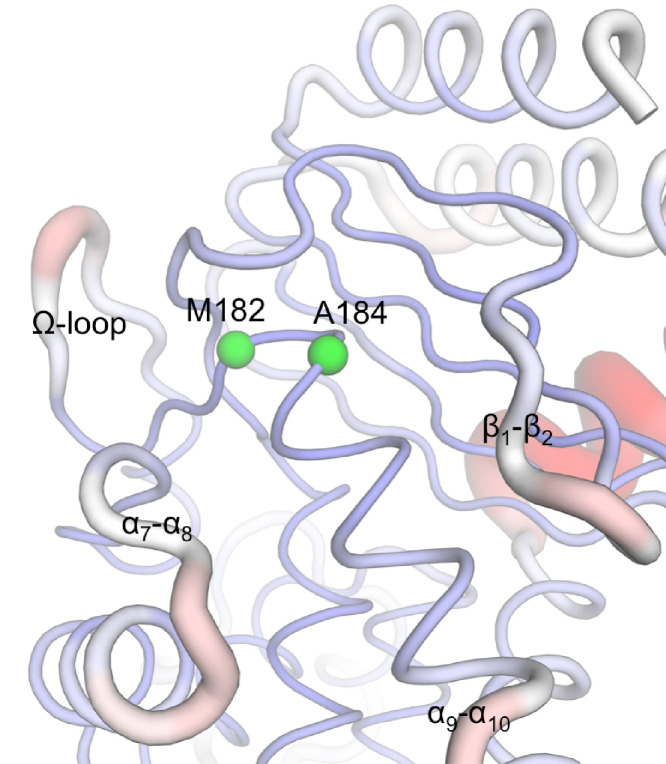
Antibiotics are crucial drugs for treating and preventing bacterial infections, but some bacteria are evolving ways to resist their effects. This ‘antibiotic resistance’ threatens lives and livelihoods worldwide. β-lactam antibiotics, like penicillin, are some of the most commonly used, but some bacteria can now make enzymes called β-lactamases, which destroy these antibiotics. Dozens of different types of β-lactamases now exist, each with different properties. Two of the most medically important are TEM-1 and KPC-2. One way to counteract β-lactamases is with drugs called inhibitors that stop the activity of these enzymes. The approved β-lactamase inhibitors work by blocking the part of the enzyme that binds and destroys antibiotics, known as the 'active site'. The β-lactamases have evolved, some of which have the ability to resist the effects of known inhibitors. It is possible that targeting parts of β-lactamases far from the active site, known as 'allosteric sites', might get around these new bacterial defences. A molecule that binds to an allosteric site might alter the enzyme's shape, or restrict its movement, making it unable to do its job. Galdadas, Qu et al. used simulations to understand how molecules binding at allosteric sites affect enzyme movement. The experiments examined the structures of both TEM-1 and KPC-2, looking at how their shapes changed as molecules were removed from the allosteric site. This revealed how the allosteric sites and the active site are linked together. When molecules were taken out of the allosteric sites, they triggered ripples of shape change that travelled via loop-like structures across the surface of the enzyme. These loops contain over half of the known differences between the different types of β-lactamases, suggesting mutations here may be responsible for changing which antibiotics each enzyme can destroy. In other words, changes in the 'ripples' may be related to the ability of the enzymes to resist particular antibiotics. Understanding how changes in one part of a β-lactamase enzyme reach the active site could help in the design of new inhibitors. It might also help to explain how β-lactamases evolve new properties. Further work could show why different enzymes are more or less active against different antibiotics.
© 2021, Galdadas et al.
Conflict of interest statement
IG, SQ, AO, EO, AM, MM, CT, FG, JS, RB, AM, SH No competing interests declared, PA Pratul K Agarwal is the founder and owner of Arium BioLabs LLC.
Figures
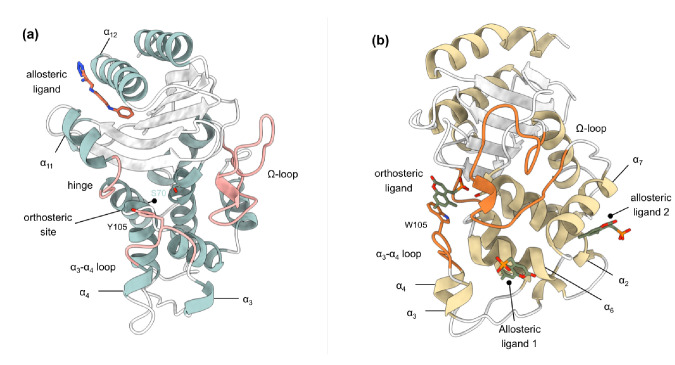
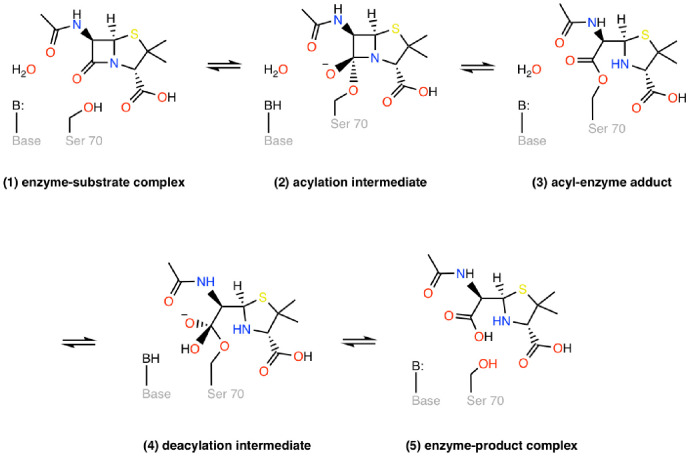
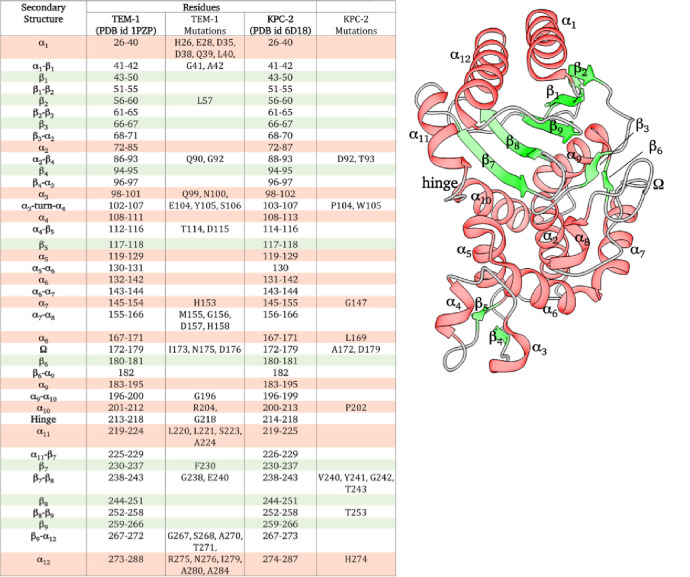
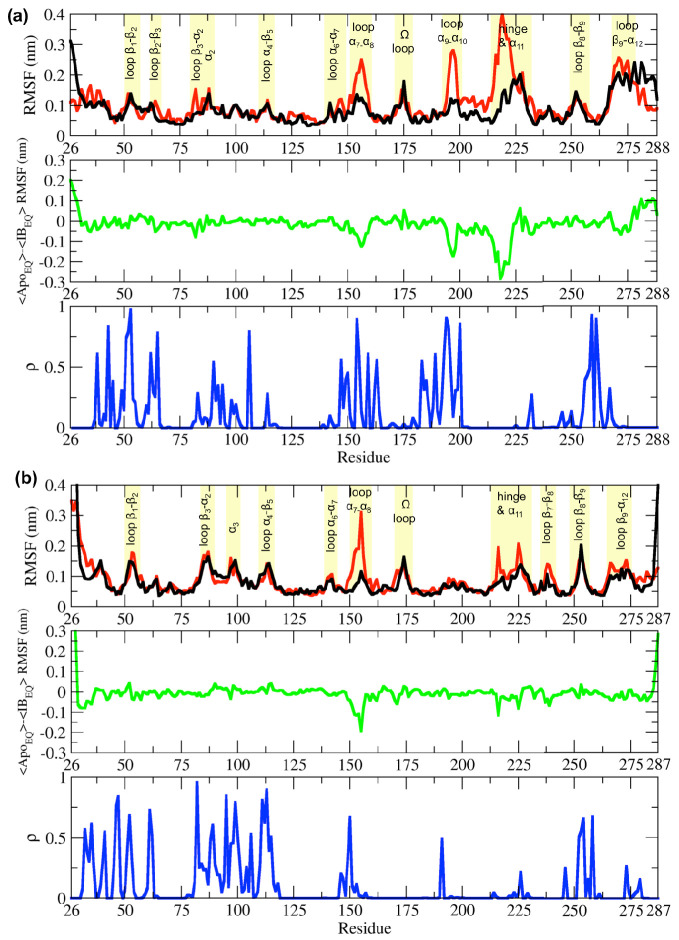
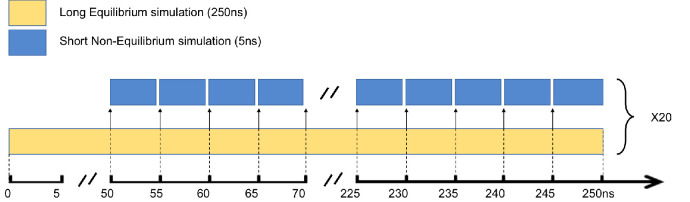
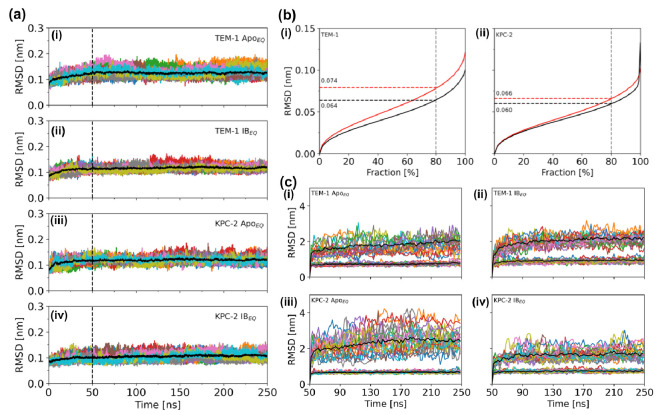
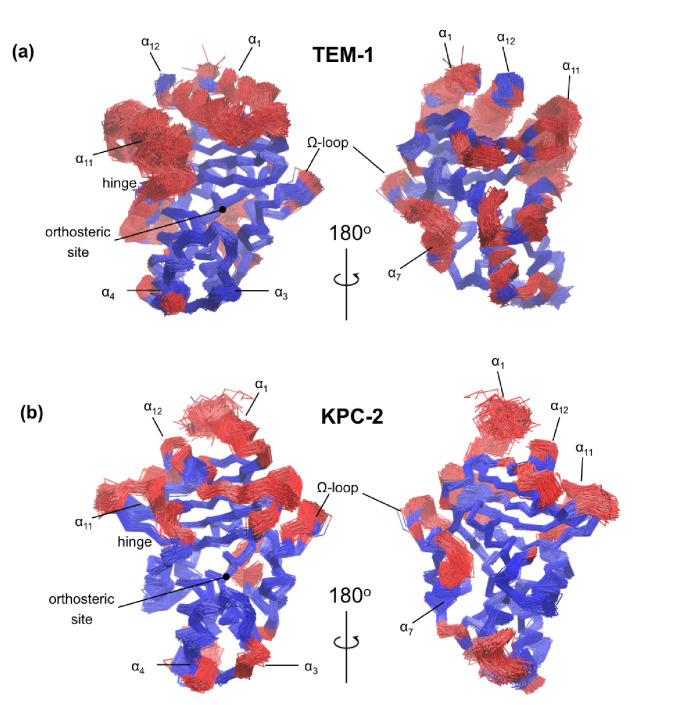
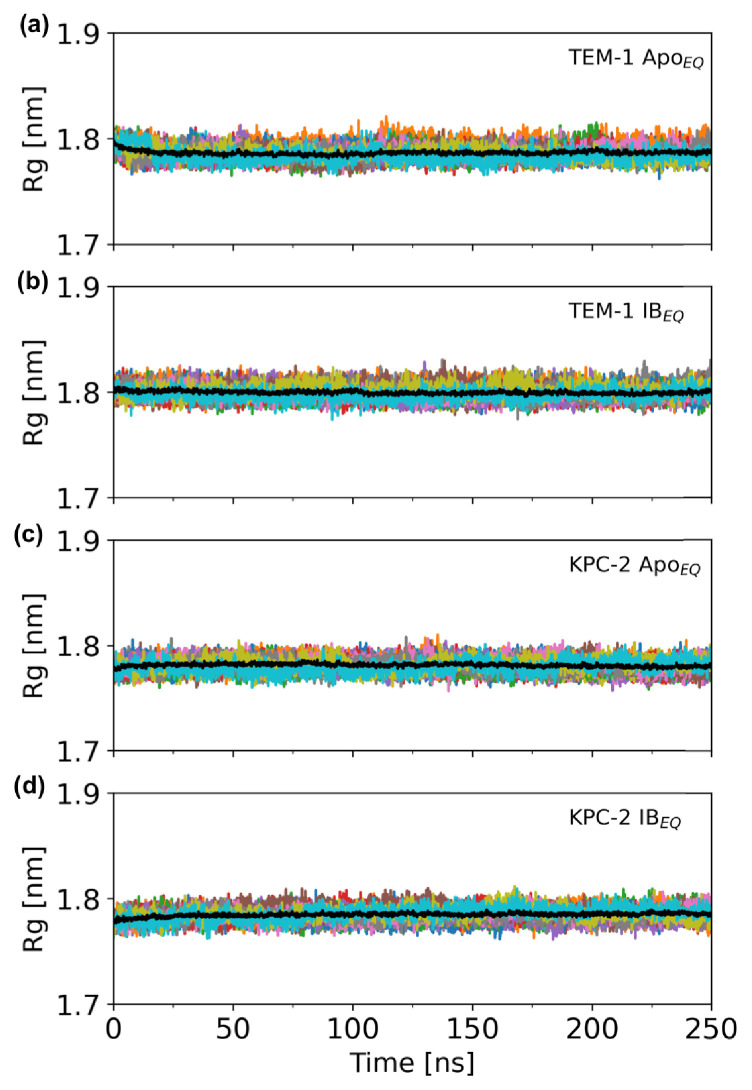
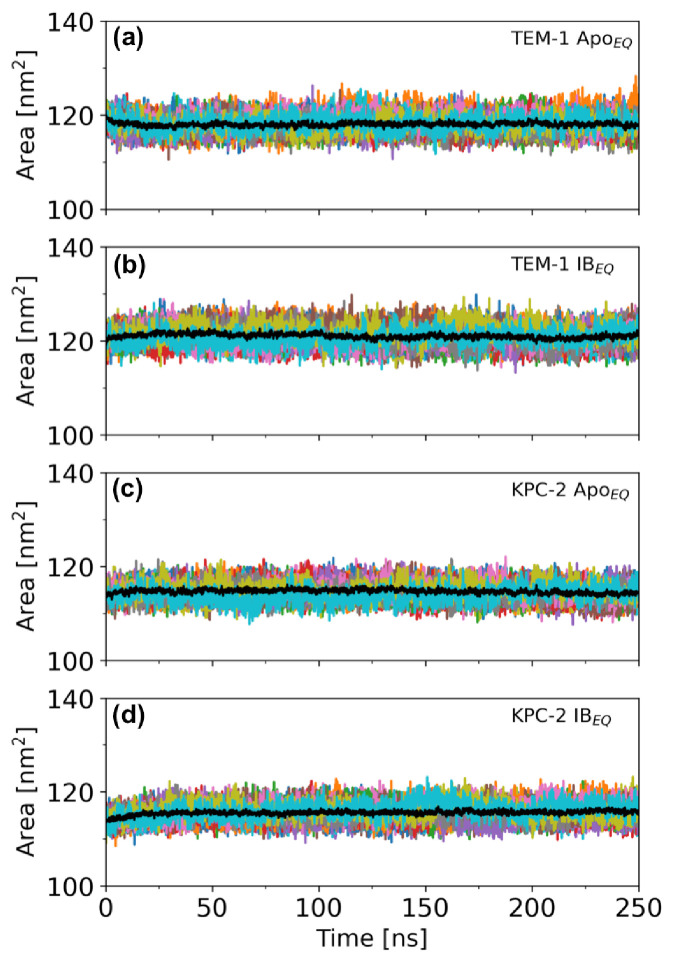
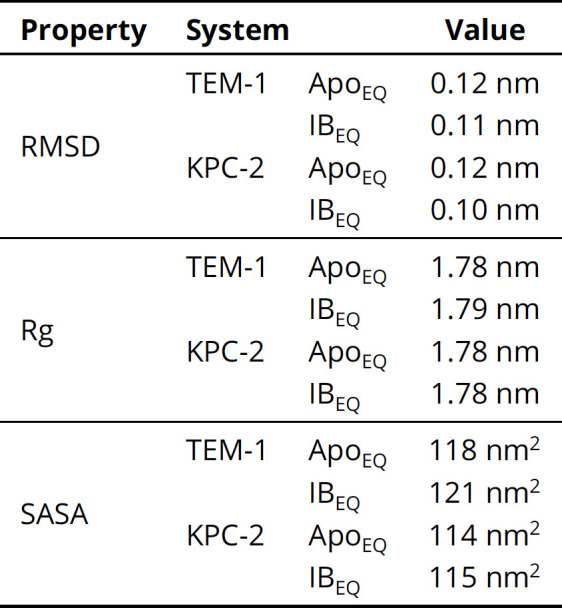

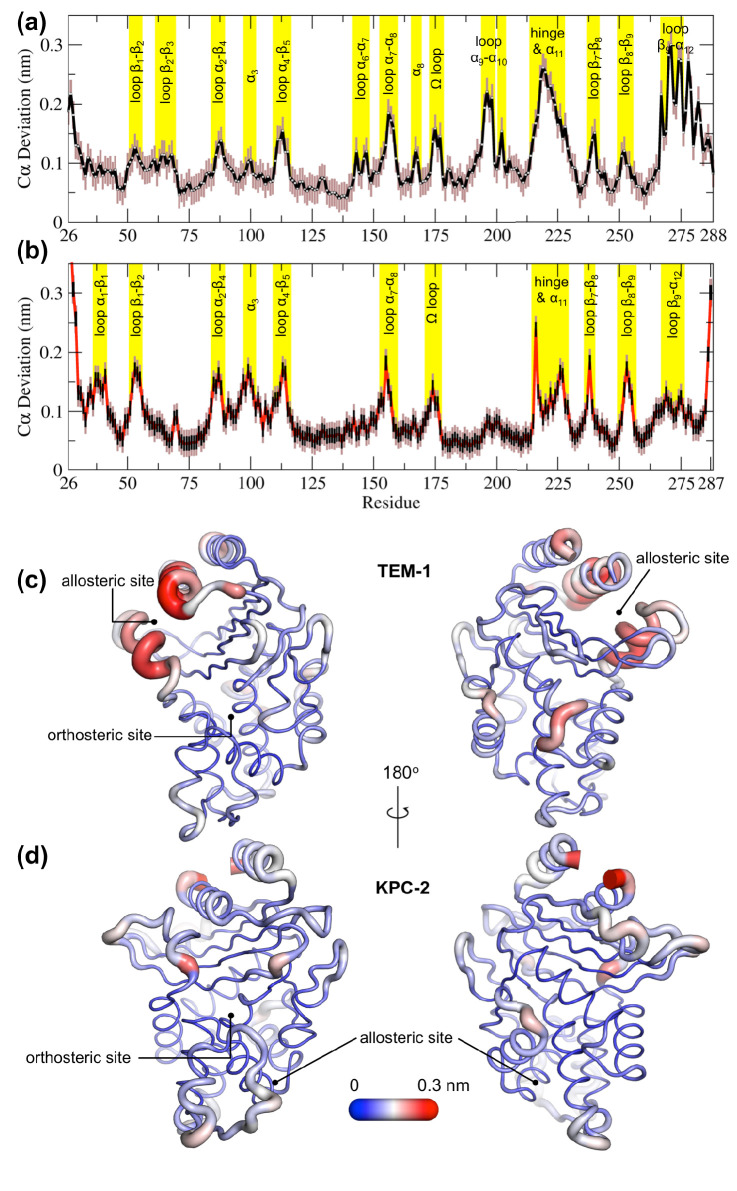
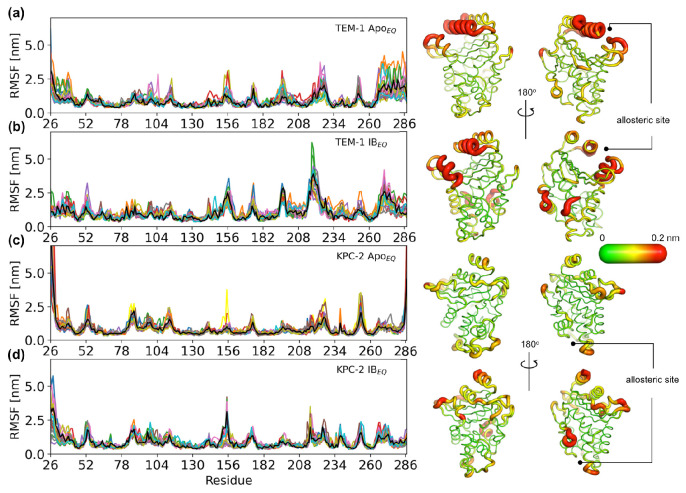
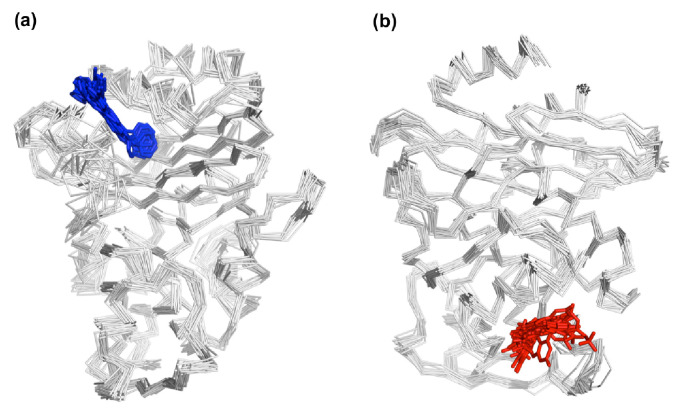
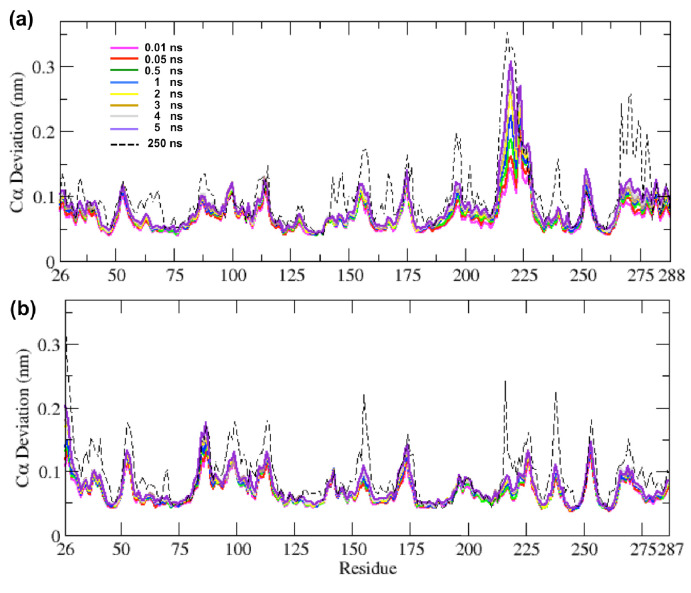
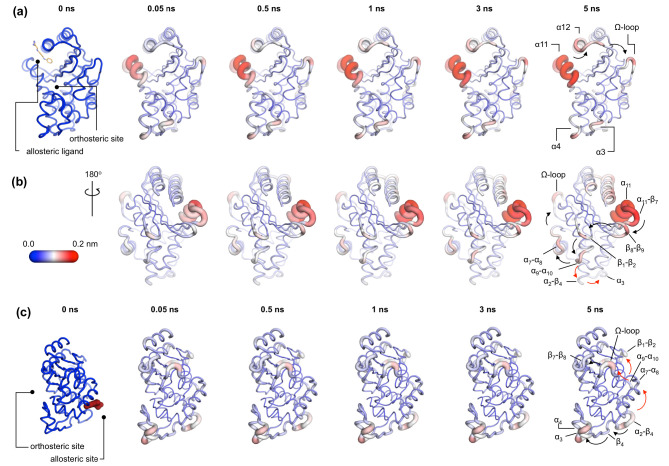
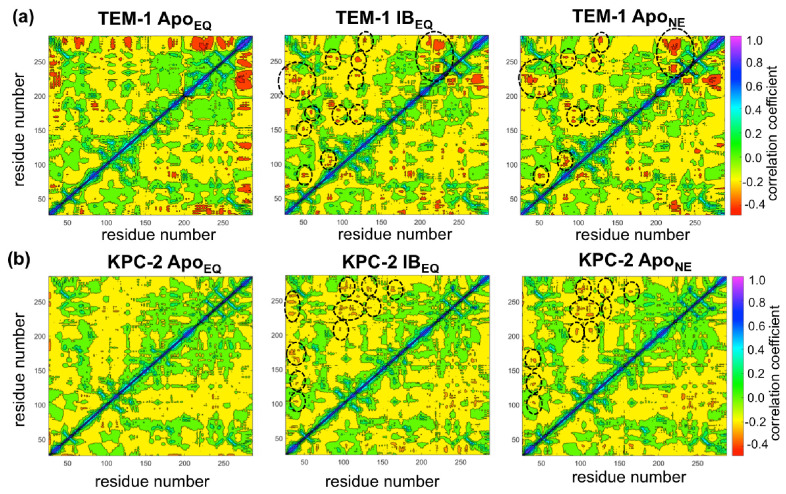
References
-
- Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, Lindahl E. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX. 2015;1-2:19–25. doi: 10.1016/j.softx.2015.06.001. - DOI
-
- Agarwal PK, Schultz C, Kalivretenos A, Ghosh B, Broedel SE. Engineering a Hyper-catalytic enzyme by photoactivated conformation modulation. The Journal of Physical Chemistry Letters. 2012;3:1142–1146. doi: 10.1021/jz201675m. - DOI
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
