

Higher infectivity of the SARS-CoV-2 new variants is associated with K417N/T, E484K, and N501Y mutants: An insight from structural data
- PMID: 33755190
- PMCID: PMC8251074
- DOI: 10.1002/jcp.30367
Higher infectivity of the SARS-CoV-2 new variants is associated with K417N/T, E484K, and N501Y mutants: An insight from structural data
Abstract
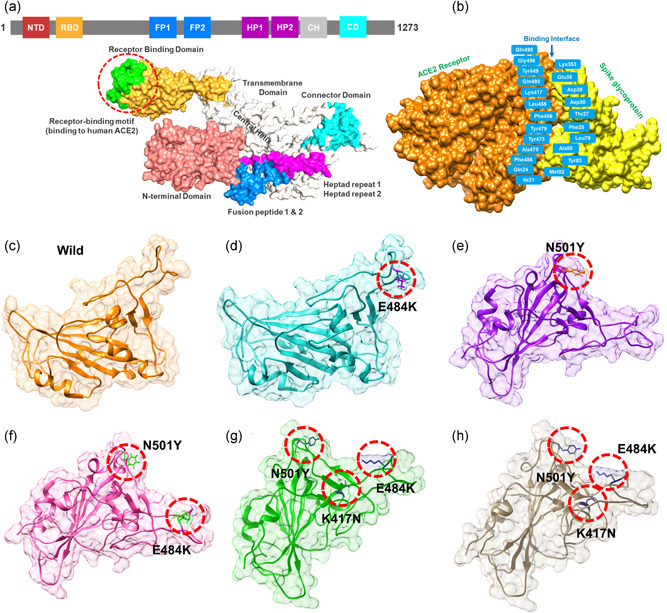
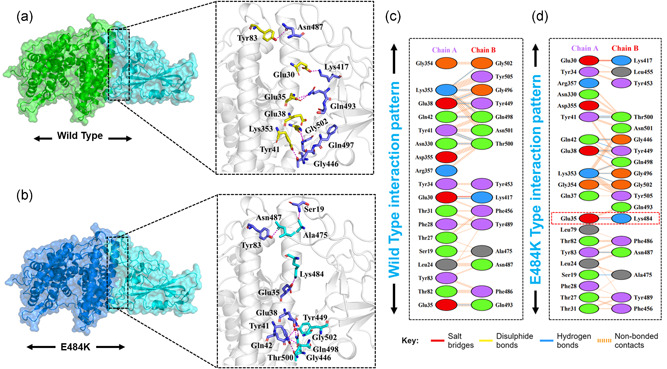
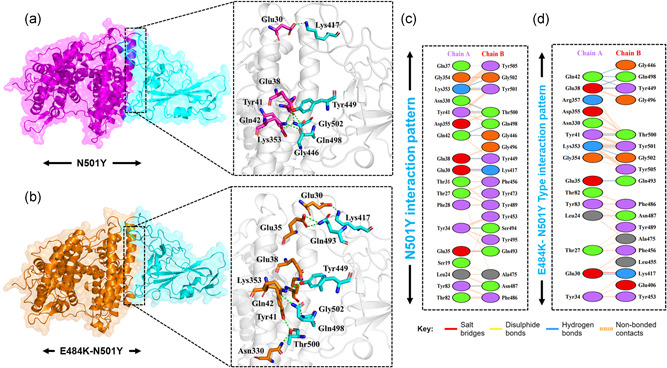
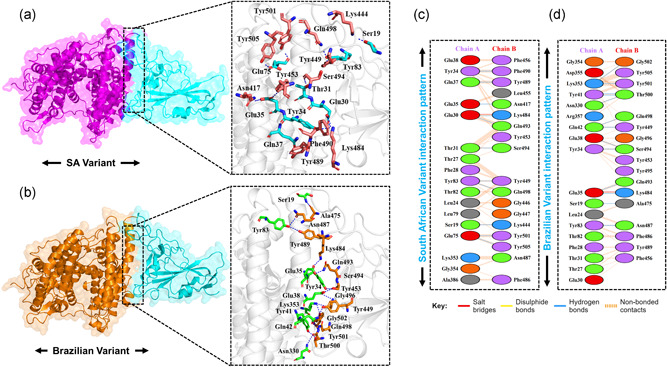
The evolution of the SARS-CoV-2 new variants reported to be 70% more contagious than the earlier one is now spreading fast worldwide. There is an instant need to discover how the new variants interact with the host receptor (ACE2). Among the reported mutations in the Spike glycoprotein of the new variants, three are specific to the receptor-binding domain (RBD) and required insightful scrutiny for new therapeutic options. These structural evolutions in the RBD domain may impart a critical role to the unique pathogenicity of the SARS-CoV-2 new variants. Herein, using structural and biophysical approaches, we explored that the specific mutations in the UK (N501Y), South African (K417N-E484K-N501Y), Brazilian (K417T-E484K-N501Y), and hypothetical (N501Y-E484K) variants alter the binding affinity, create new inter-protein contacts and changes the internal structural dynamics thereby increases the binding and eventually the infectivity. Our investigation highlighted that the South African (K417N-E484K-N501Y), Brazilian (K417T-E484K-N501Y) variants are more lethal than the UK variant (N501Y). The behavior of the wild type and N501Y is comparable. Free energy calculations further confirmed that increased binding of the spike RBD to the ACE2 is mainly due to the electrostatic contribution. Further, we find that the unusual virulence of this virus is potentially the consequence of Darwinian selection-driven epistasis in protein evolution. The triple mutants (South African and Brazilian) may pose a serious threat to the efficacy of the already developed vaccine. Our analysis would help to understand the binding and structural dynamics of the new mutations in the RBD domain of the Spike protein and demand further investigation in in vitro and in vivo models to design potential therapeutics against the new variants.
Keywords: KD (dissociation constant); MD simulation; SARS-CoV-2; new variants; protein-protein docking.
© 2021 Wiley Periodicals LLC.
Conflict of interest statement
The authors declare that there are no conflict of interests.
Figures
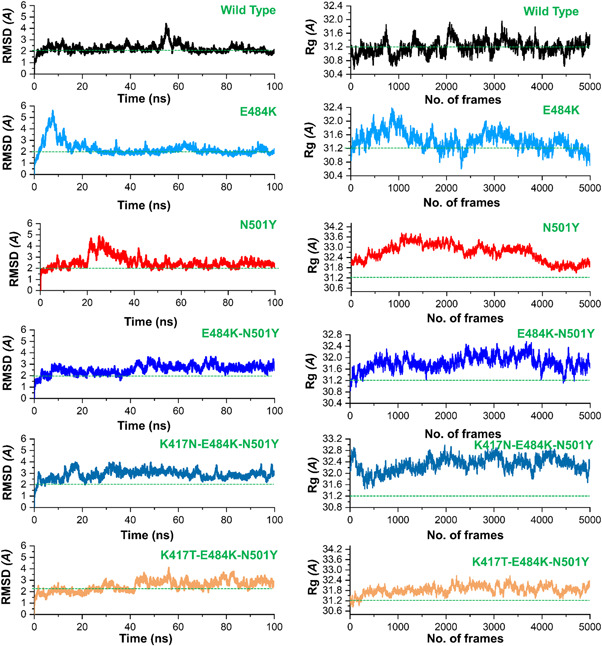
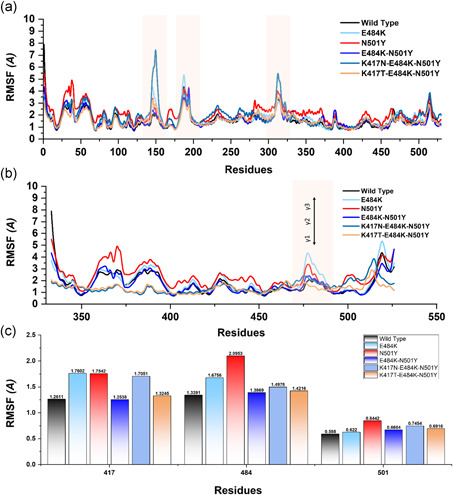
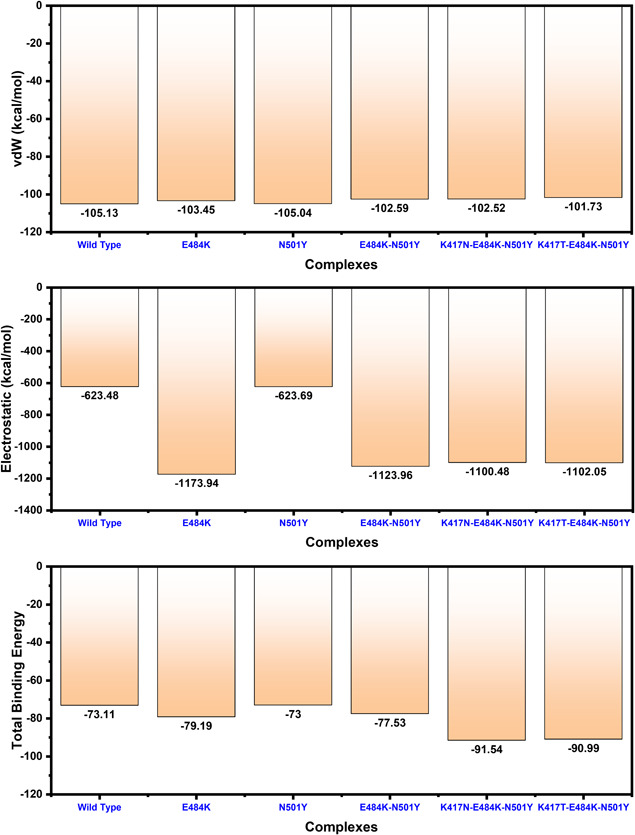
References
-
- Ali, A. , Khan, A. , Kaushik, A. C. , Wang, Y. , Ali, S. S. , Junaid, M. , Saleem, S. , Cho, W. C. , Mao, X. , & Wei, D.‐Q. (2019). Immunoinformatic and systems biology approaches to predict and validate peptide vaccines against Epstein–Barr virus (EBV). Scientific Reports, 9(1), 1–12. - PMC - PubMed
-
- Davies, N. G. , Barnard, R. C. , Jarvis, C. I. , Kucharski, A. J. , Munday, J. , Pearson, C. A. , Russell, T. W. , Tully, D. C. , Abbott, S. , & Gimma, A. (2020). Estimated transmissibility and severity of novel SARS‐CoV‐2 Variant of Concern 202012/01 in England. medRxiv.
-
- Dominguez, C. , Boelens, R. , & Bonvin, A. M. (2003). HADDOCK: A protein−protein docking approach based on biochemical or biophysical information. Journal of the American Chemical Society, 125(7), 1731–1737. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
