

Activation of the GPR35 pathway drives angiogenesis in the tumour microenvironment
- PMID: 33758004
- PMCID: PMC8862021
- DOI: 10.1136/gutjnl-2020-323363
Activation of the GPR35 pathway drives angiogenesis in the tumour microenvironment
Abstract
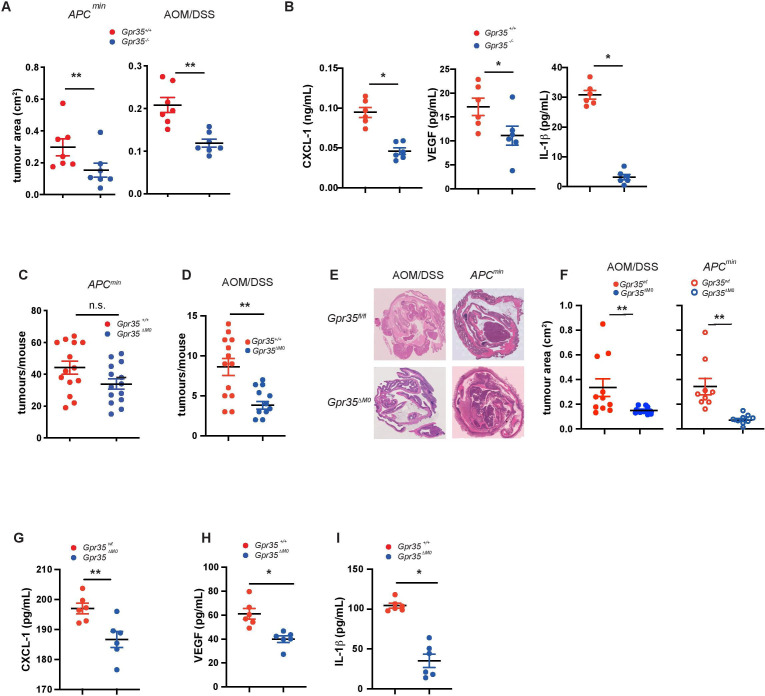
Objective: Primary sclerosing cholangitis (PSC) is in 70% of cases associated with inflammatory bowel disease. The hypermorphic T108M variant of the orphan G protein-coupled receptor GPR35 increases risk for PSC and ulcerative colitis (UC), conditions strongly predisposing for inflammation-associated liver and colon cancer. Lack of GPR35 reduces tumour numbers in mouse models of spontaneous and colitis associated cancer. The tumour microenvironment substantially determines tumour growth, and tumour-associated macrophages are crucial for neovascularisation. We aim to understand the role of the GPR35 pathway in the tumour microenvironment of spontaneous and colitis-associated colon cancers.
Design: Mice lacking GPR35 on their macrophages underwent models of spontaneous colon cancer or colitis-associated cancer. The role of tumour-associated macrophages was then assessed in biochemical and functional assays.
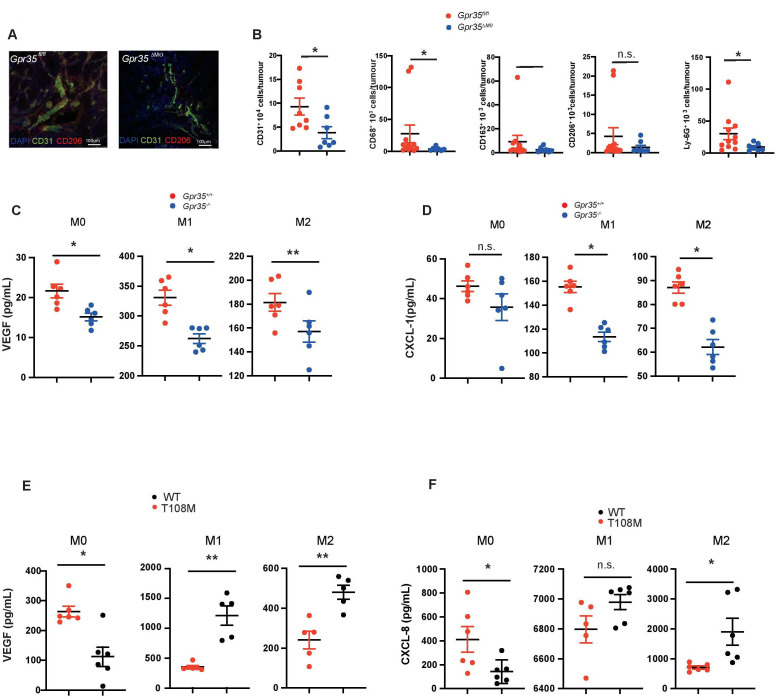
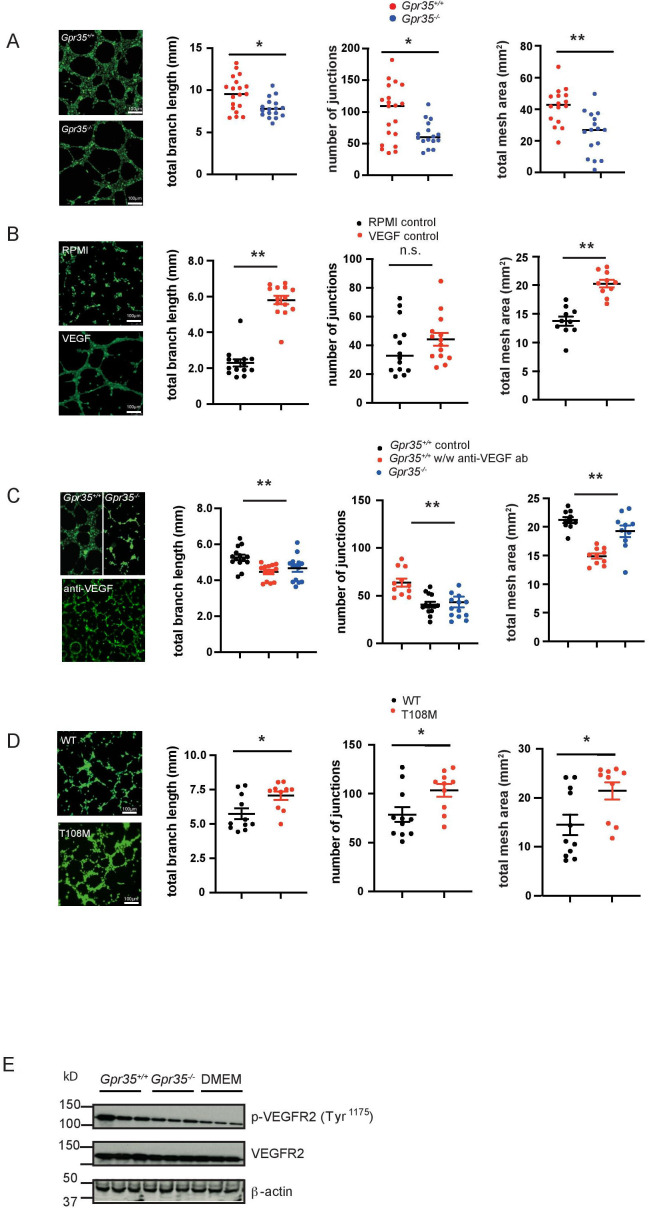
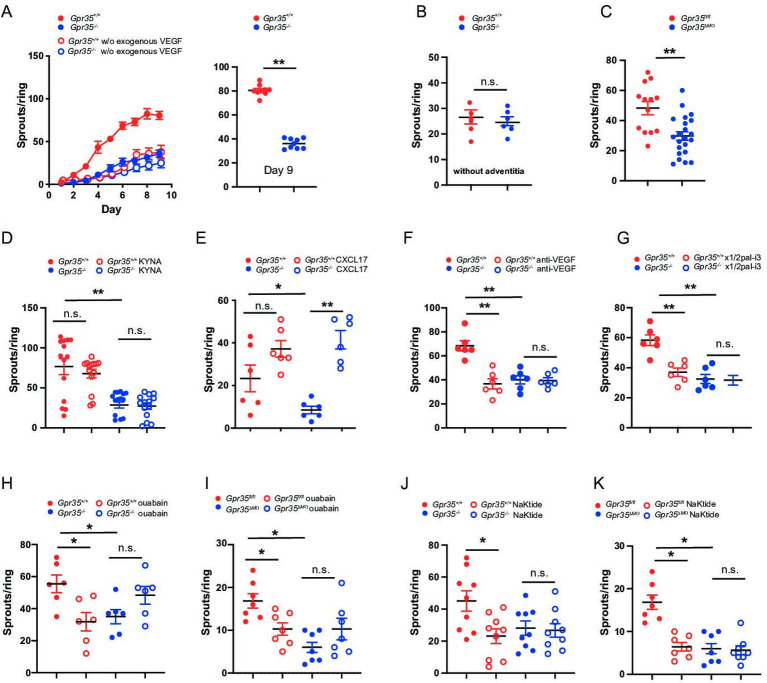
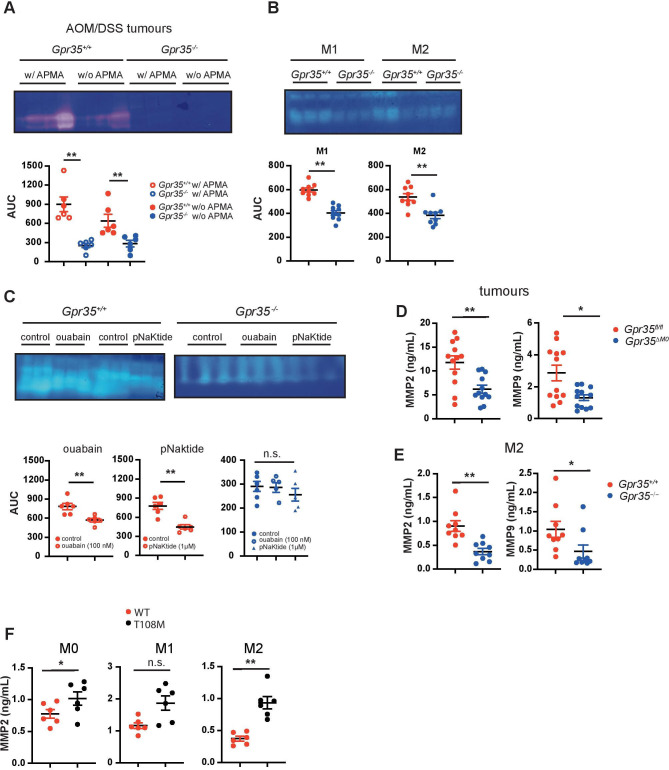
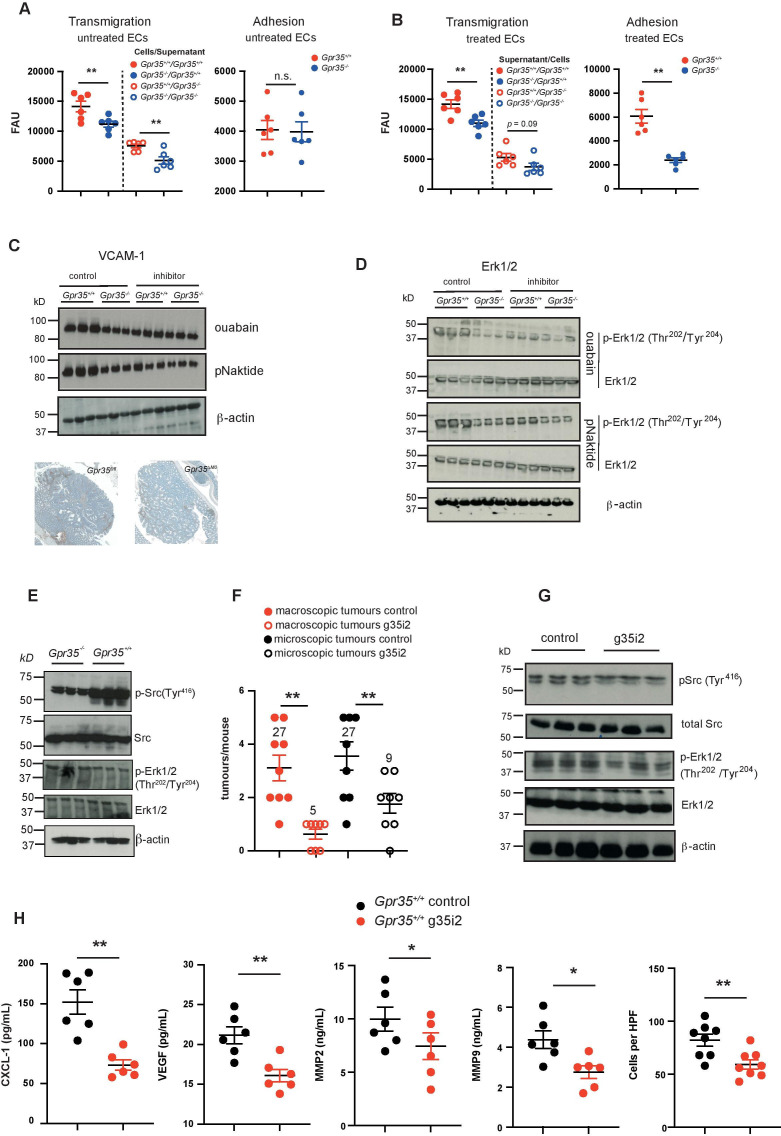
Results: Here, we show that GPR35 on macrophages is a potent amplifier of tumour growth by stimulating neoangiogenesis and tumour tissue remodelling. Deletion of Gpr35 in macrophages profoundly reduces tumour growth in inflammation-associated and spontaneous tumour models caused by mutant tumour suppressor adenomatous polyposis coli. Neoangiogenesis and matrix metalloproteinase activity is promoted by GPR35 via Na/K-ATPase-dependent ion pumping and Src activation, and is selectively inhibited by a GPR35-specific pepducin. Supernatants from human inducible-pluripotent-stem-cell derived macrophages carrying the UC and PSC risk variant stimulate tube formation by enhancing the release of angiogenic factors.
Conclusions: Activation of the GPR35 pathway promotes tumour growth via two separate routes, by directly augmenting proliferation in epithelial cells that express the receptor, and by coordinating macrophages' ability to create a tumour-permissive environment.
Keywords: angiogenesis; colorectal cancer; primary sclerosing cholangitis; receptor characterisation; ulcerative colitis.
© Author(s) (or their employer(s)) 2022. Re-use permitted under CC BY. Published by BMJ.
Conflict of interest statement
Competing interests: None declared.
Figures
Similar articles
-
GPR35 promotes glycolysis, proliferation, and oncogenic signaling by engaging with the sodium potassium pump.Sci Signal. 2019 Jan 1;12(562):eaau9048. doi: 10.1126/scisignal.aau9048. Sci Signal. 2019. PMID: 30600262 Free PMC article.
-
Genome-wide association analysis in primary sclerosing cholangitis and ulcerative colitis identifies risk loci at GPR35 and TCF4.Hepatology. 2013 Sep;58(3):1074-83. doi: 10.1002/hep.25977. Epub 2013 Jan 17. Hepatology. 2013. PMID: 22821403
-
Natural history of low grade dysplasia in patients with primary sclerosing cholangitis and ulcerative colitis.J Crohns Colitis. 2013 Dec;7(12):968-73. doi: 10.1016/j.crohns.2013.02.002. Epub 2013 Feb 20. J Crohns Colitis. 2013. PMID: 23433613
-
Inflammatory bowel disease of primary sclerosing cholangitis: a distinct entity?World J Gastroenterol. 2014 Mar 28;20(12):3245-54. doi: 10.3748/wjg.v20.i12.3245. World J Gastroenterol. 2014. PMID: 24696608 Free PMC article. Review.
-
[Primary sclerosing cholangitis--an ulcerative colitis-associated illness with surgical consequences].Chirurg. 1998 Oct;69(10):1028-34. Chirurg. 1998. PMID: 9833181 Review. German.
Cited by
-
From orphan to oncogene: The role of GPR35 in cancer and immune modulation.Cytokine Growth Factor Rev. 2024 Jun;77:56-66. doi: 10.1016/j.cytogfr.2024.03.004. Epub 2024 Mar 19. Cytokine Growth Factor Rev. 2024. PMID: 38514303 Free PMC article. Review.
-
Involvement of CXCL17 and GPR35 in Gastric Cancer Initiation and Progression.Int J Mol Sci. 2022 Dec 29;24(1):615. doi: 10.3390/ijms24010615. Int J Mol Sci. 2022. PMID: 36614059 Free PMC article.
-
Exploring oncogenes for renal clear cell carcinoma based on G protein-coupled receptor-associated genes.Discov Oncol. 2023 Oct 10;14(1):182. doi: 10.1007/s12672-023-00795-z. Discov Oncol. 2023. PMID: 37816979 Free PMC article.
-
The Role of Herb-Partitioned Moxibustion in the Angiogenesis of Colitis-Associated Cancer in Rats.J Inflamm Res. 2025 May 23;18:6623-6639. doi: 10.2147/JIR.S518214. eCollection 2025. J Inflamm Res. 2025. PMID: 40438180 Free PMC article.
-
Multi-Omics Analysis for Transcriptional Regulation of Immune-Related Targets Using Epigenetic Data: A New Research Direction.Front Immunol. 2022 Jan 3;12:741634. doi: 10.3389/fimmu.2021.741634. eCollection 2021. Front Immunol. 2022. PMID: 35046932 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous