

CellMixS: quantifying and visualizing batch effects in single-cell RNA-seq data
- PMID: 33758076
- PMCID: PMC7994321
- DOI: 10.26508/lsa.202001004
CellMixS: quantifying and visualizing batch effects in single-cell RNA-seq data
Abstract
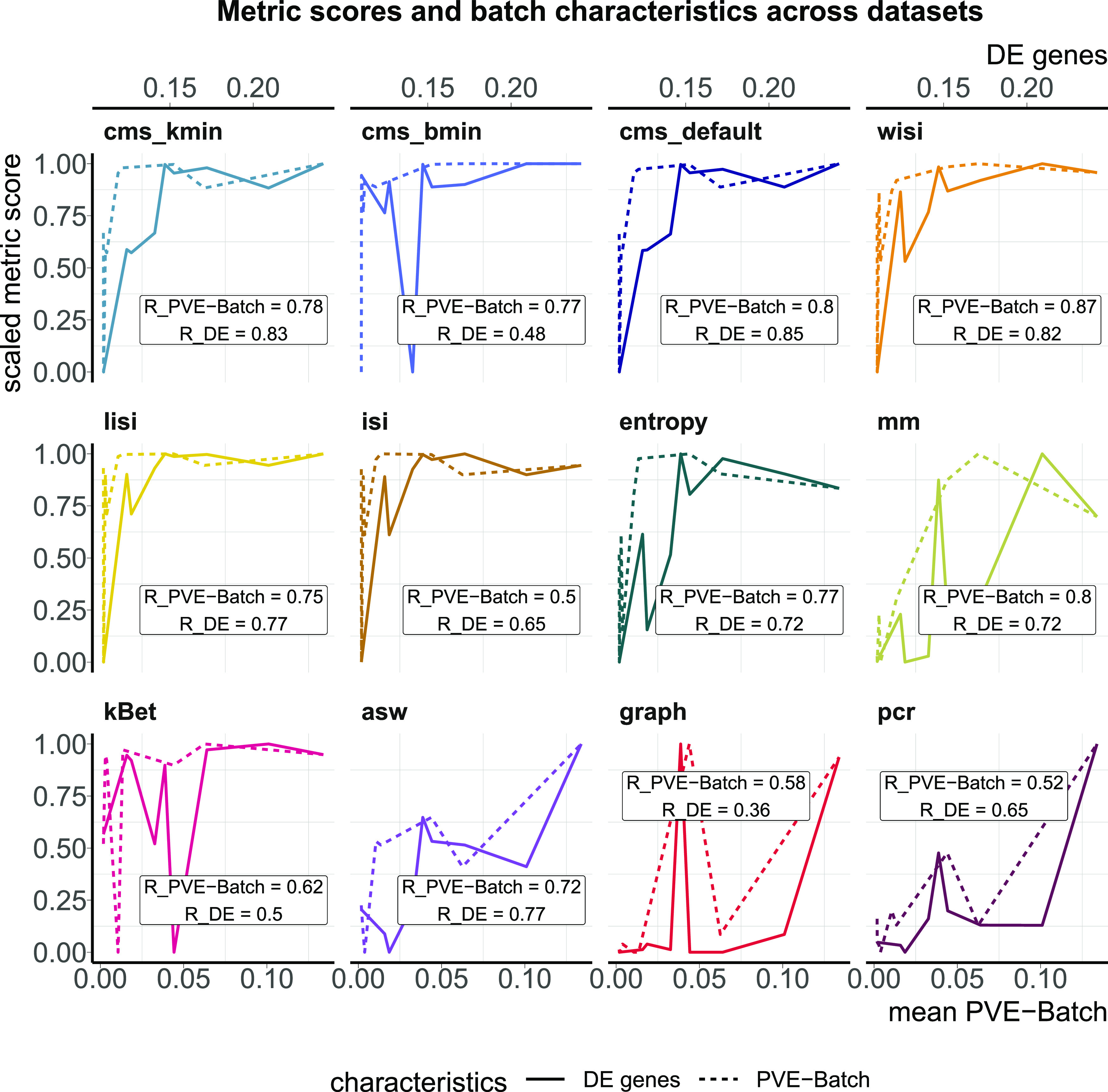
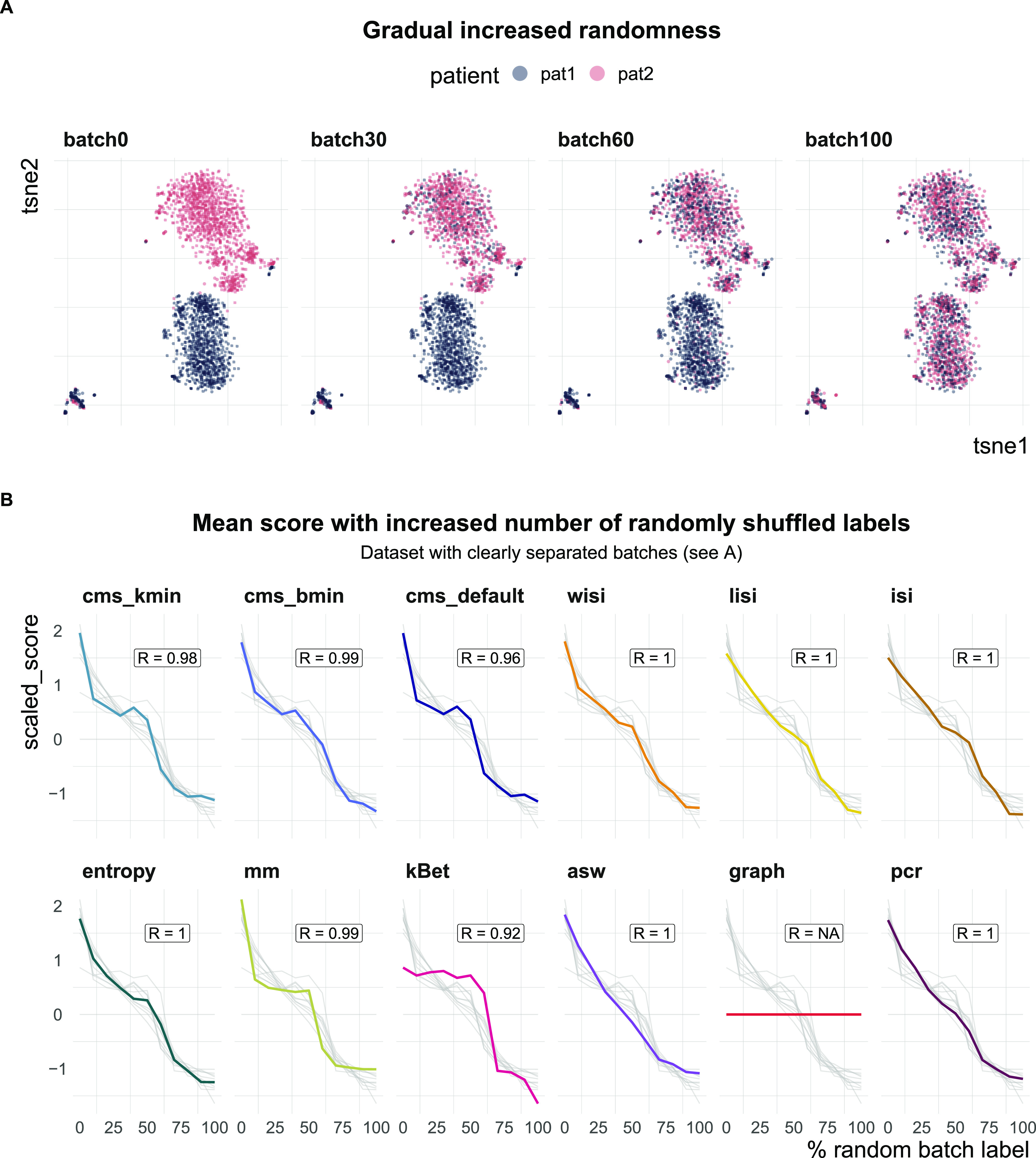
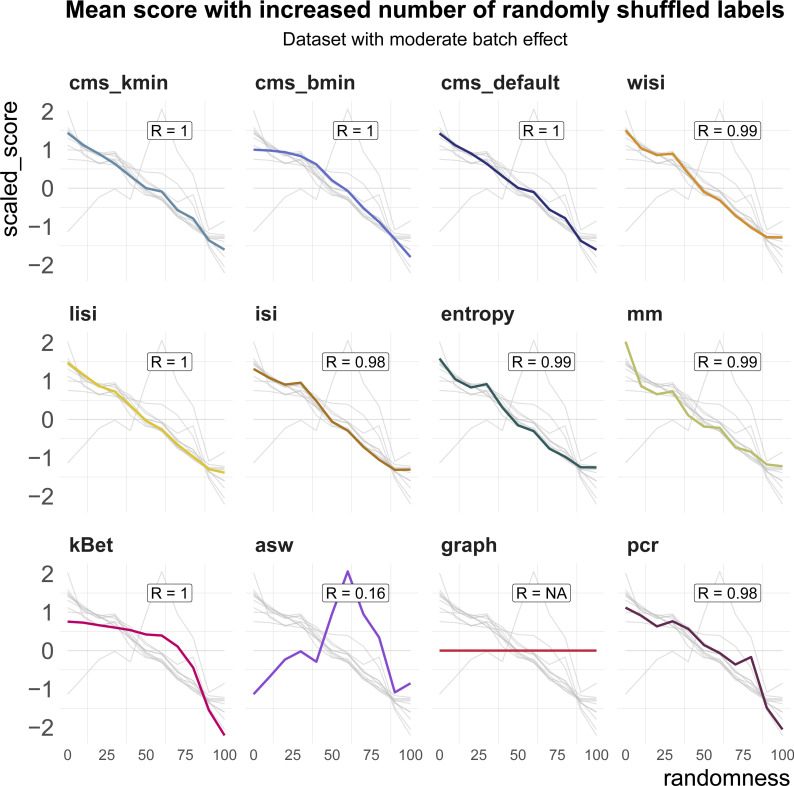
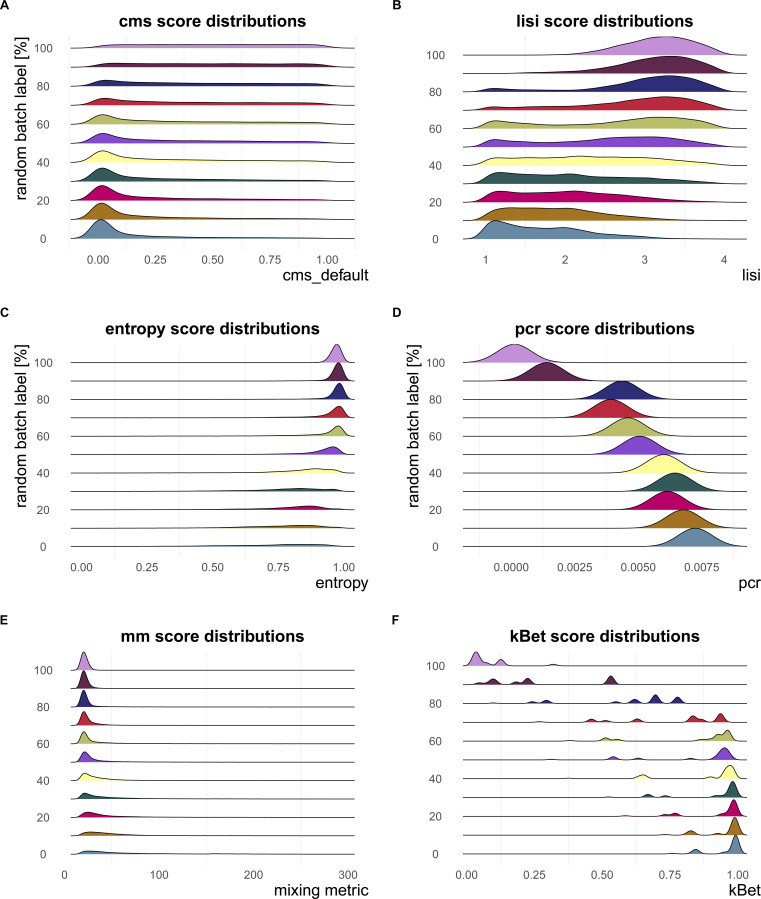
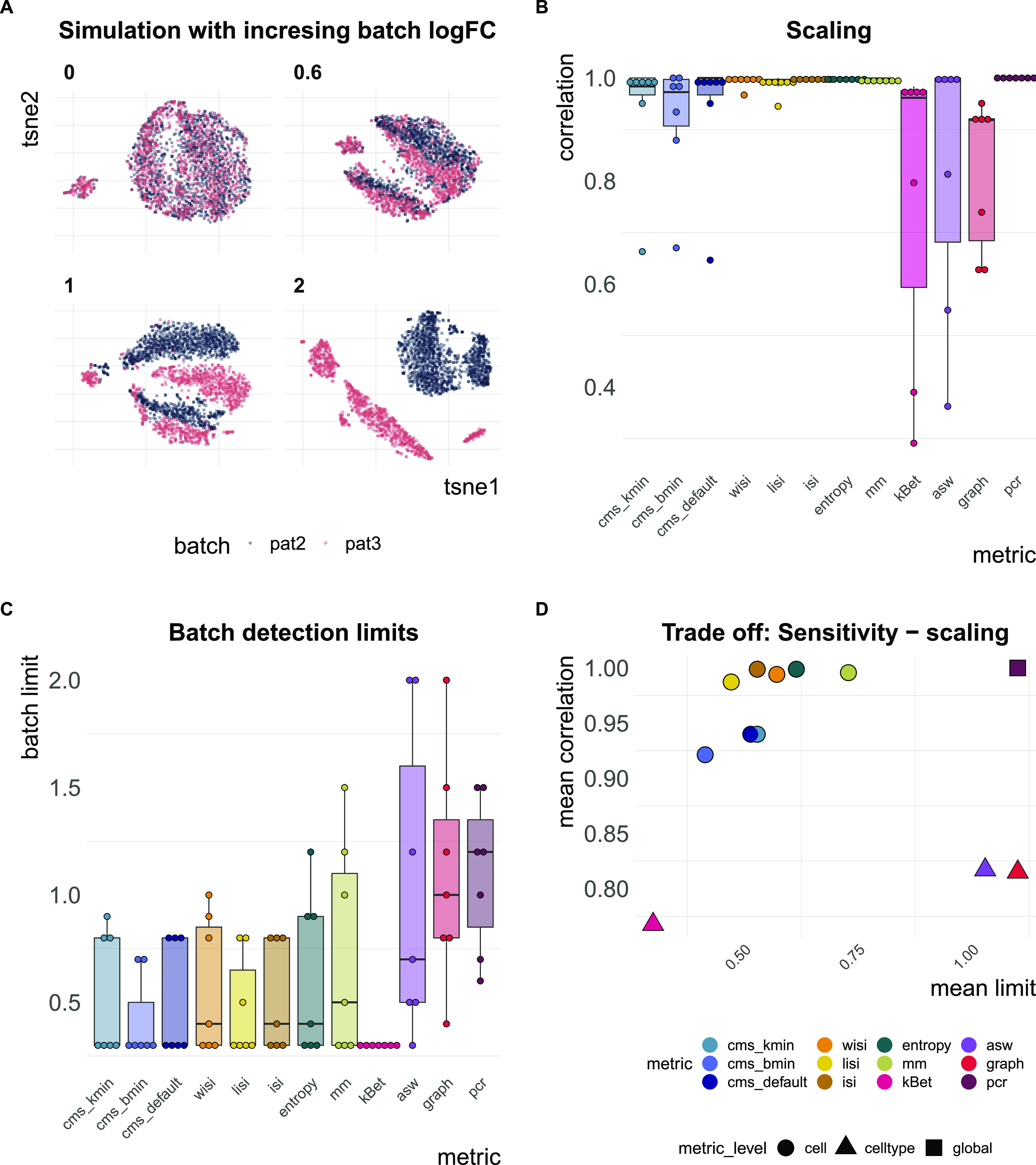
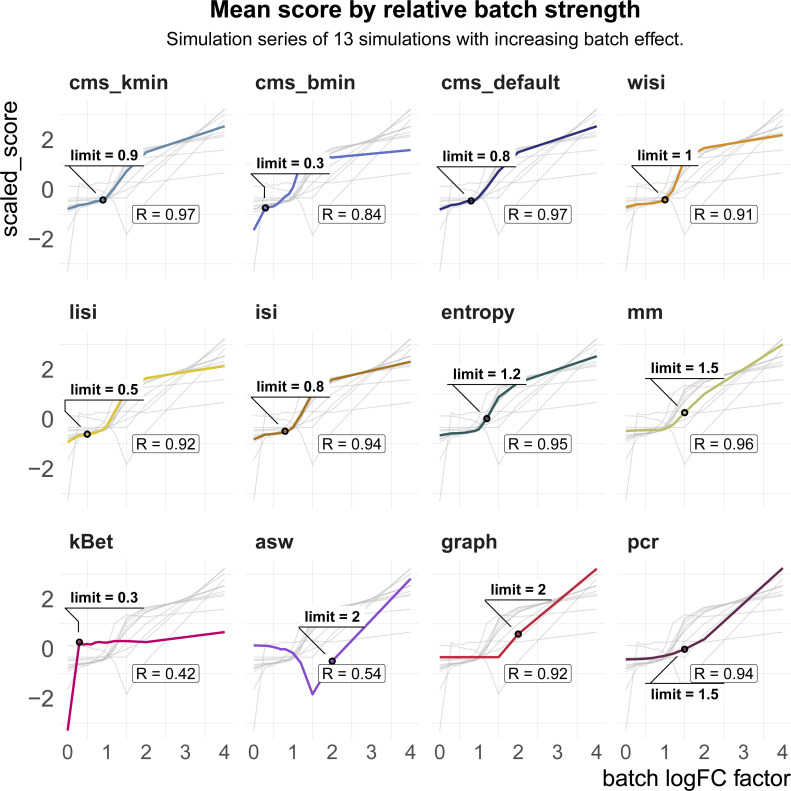
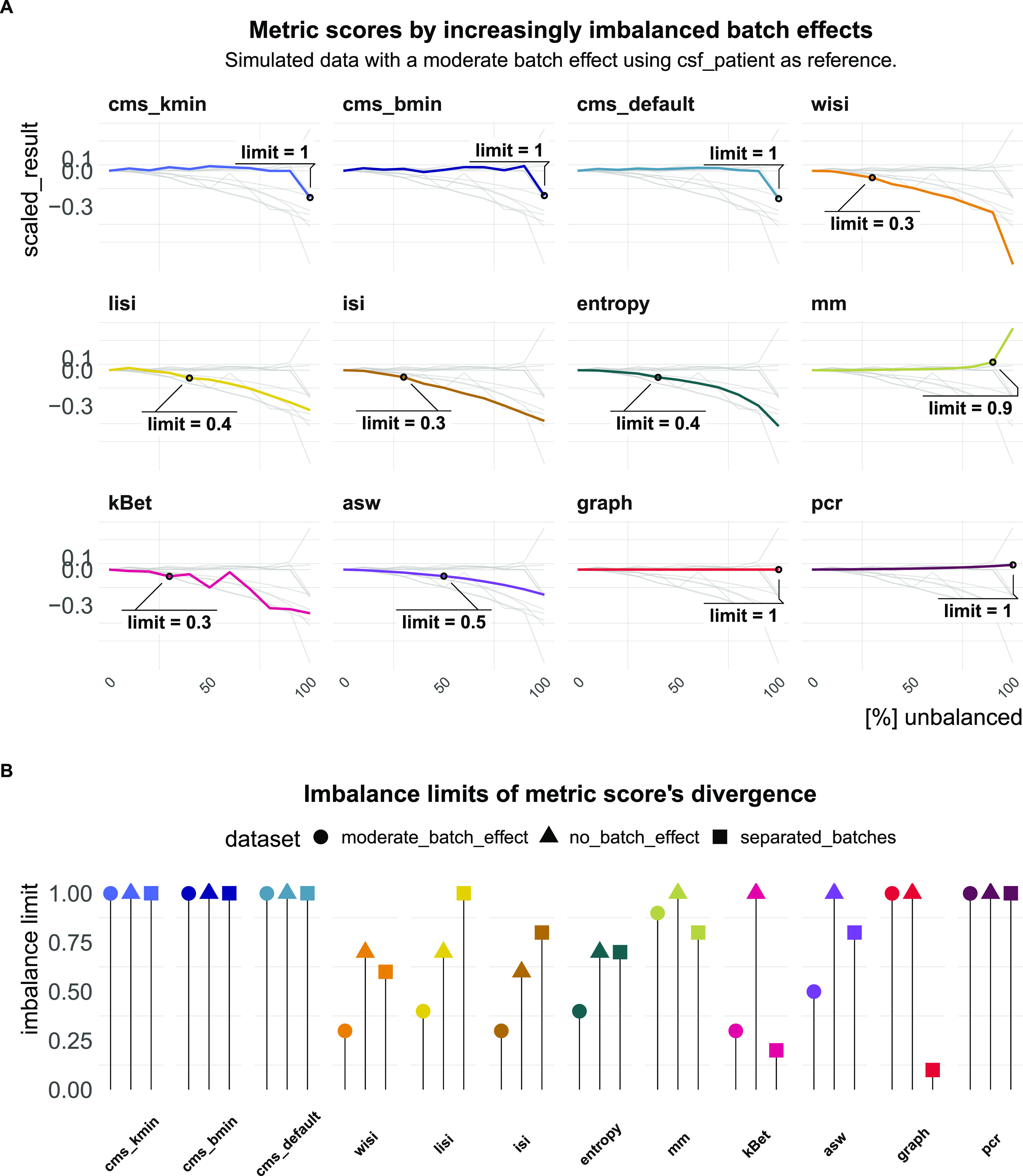
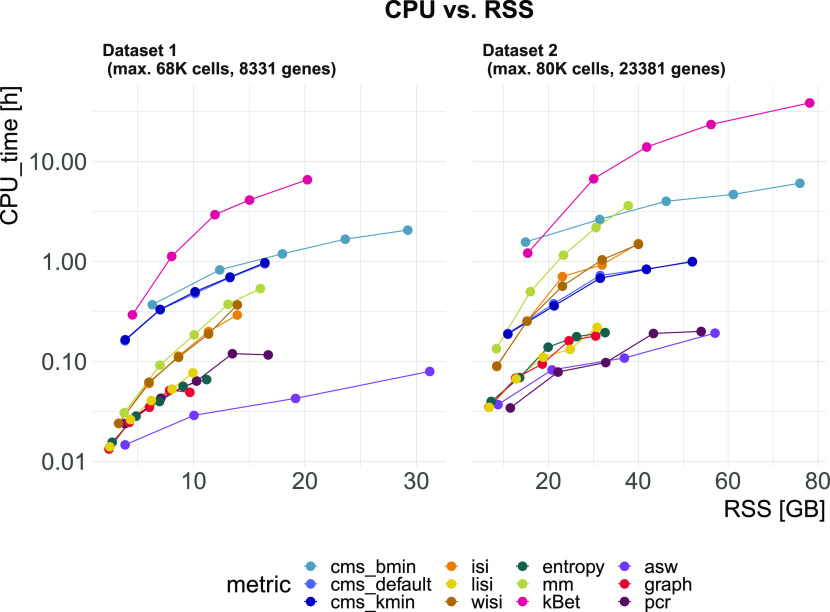
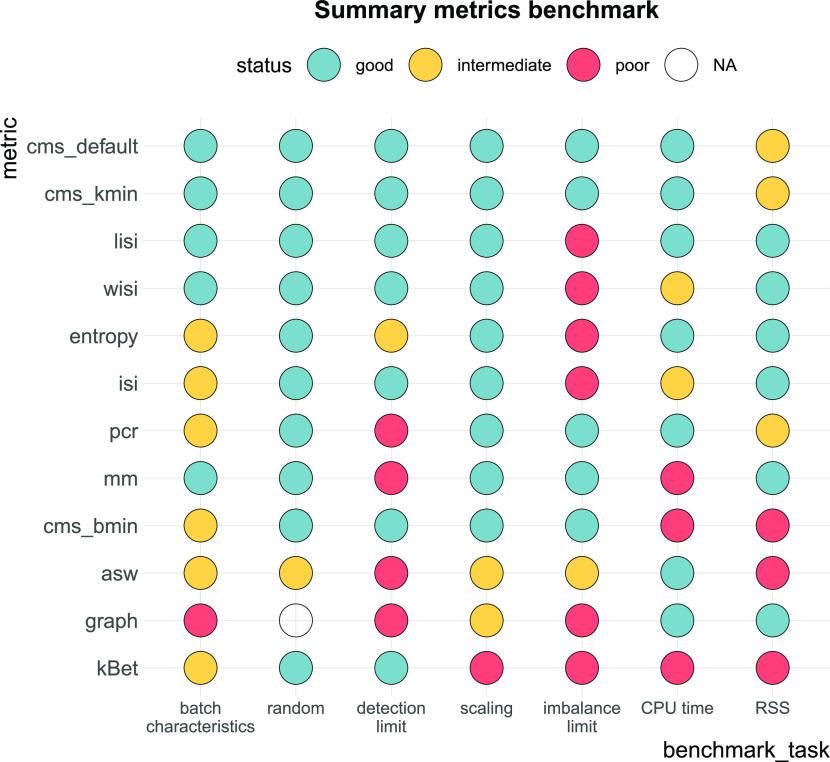
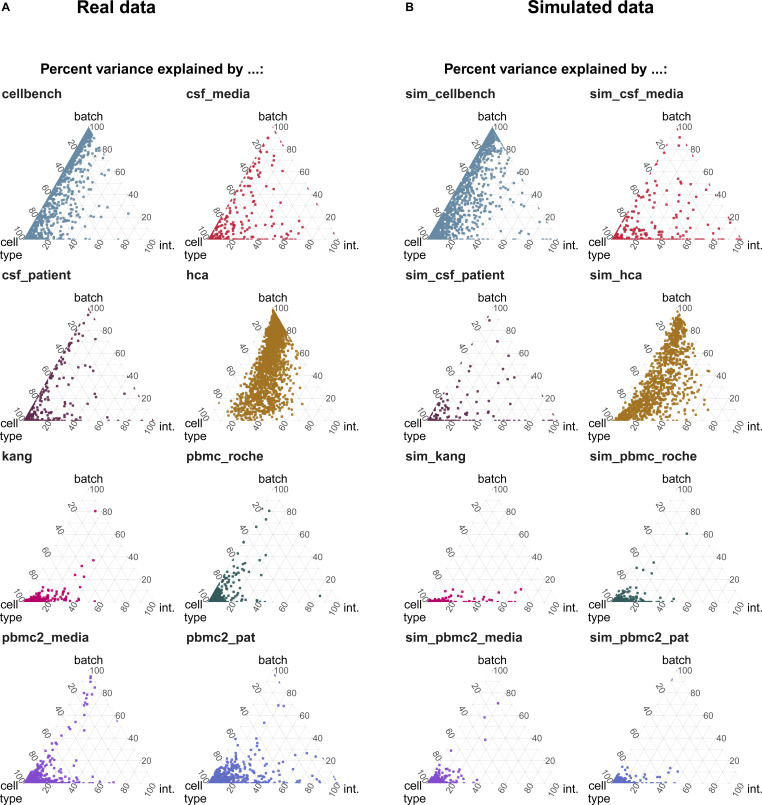
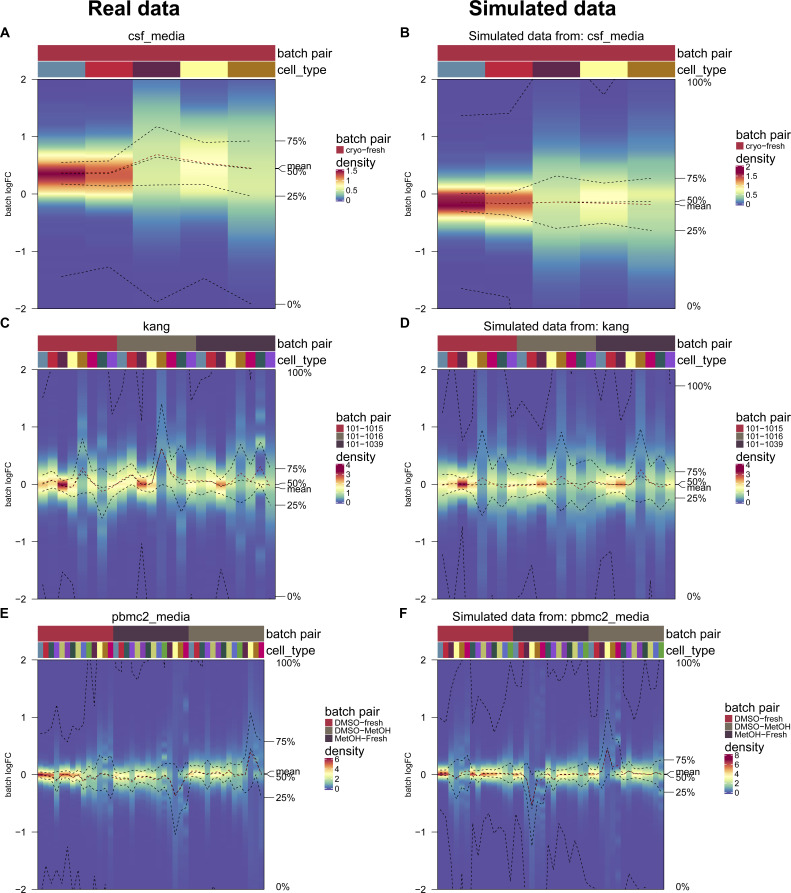
A key challenge in single-cell RNA-sequencing (scRNA-seq) data analysis is batch effects that can obscure the biological signal of interest. Although there are various tools and methods to correct for batch effects, their performance can vary. Therefore, it is important to understand how batch effects manifest to adjust for them. Here, we systematically explore batch effects across various scRNA-seq datasets according to magnitude, cell type specificity, and complexity. We developed a cell-specific mixing score (cms) that quantifies mixing of cells from multiple batches. By considering distance distributions, the score is able to detect local batch bias as well as differentiate between unbalanced batches and systematic differences between cells of the same cell type. We compare metrics in scRNA-seq data using real and synthetic datasets and whereas these metrics target the same question and are used interchangeably, we find differences in scalability, sensitivity, and ability to handle differentially abundant cell types. We find that cell-specific metrics outperform cell type-specific and global metrics and recommend them for both method benchmarks and batch exploration.
© 2021 Lütge et al.
Conflict of interest statement
D Malhotra is a full-time employee of Roche; the remaining authors declare that they have no competing interests.
Figures
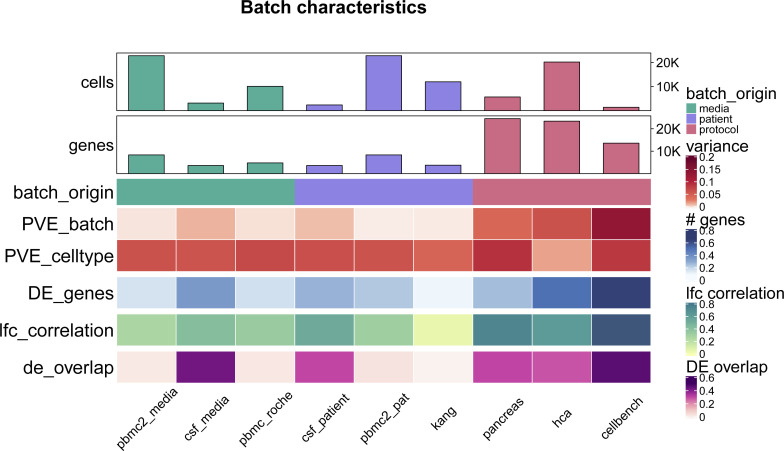
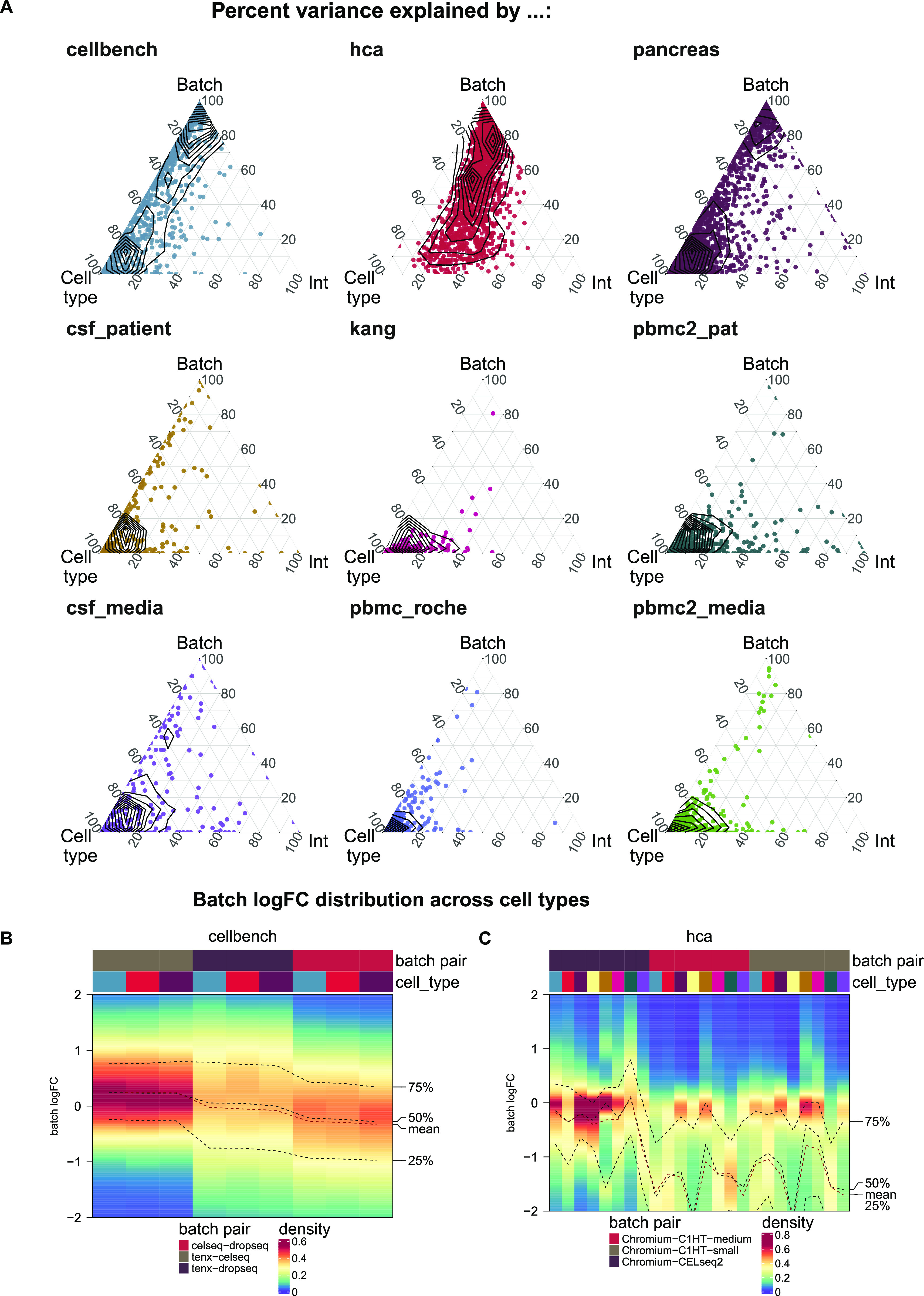
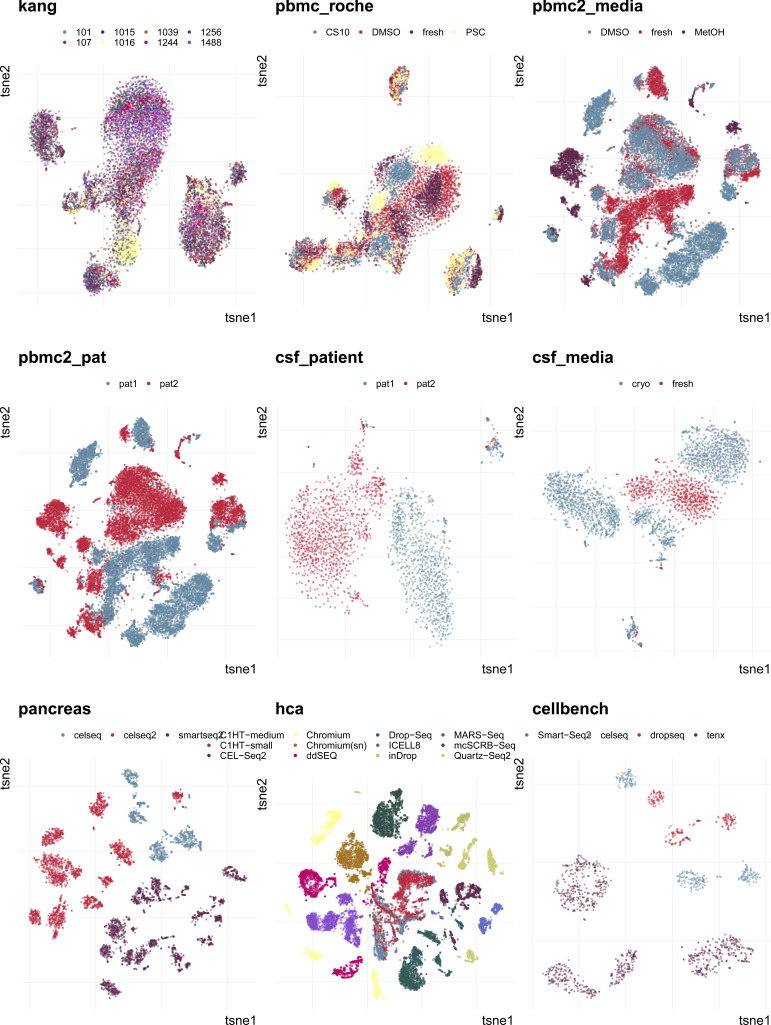
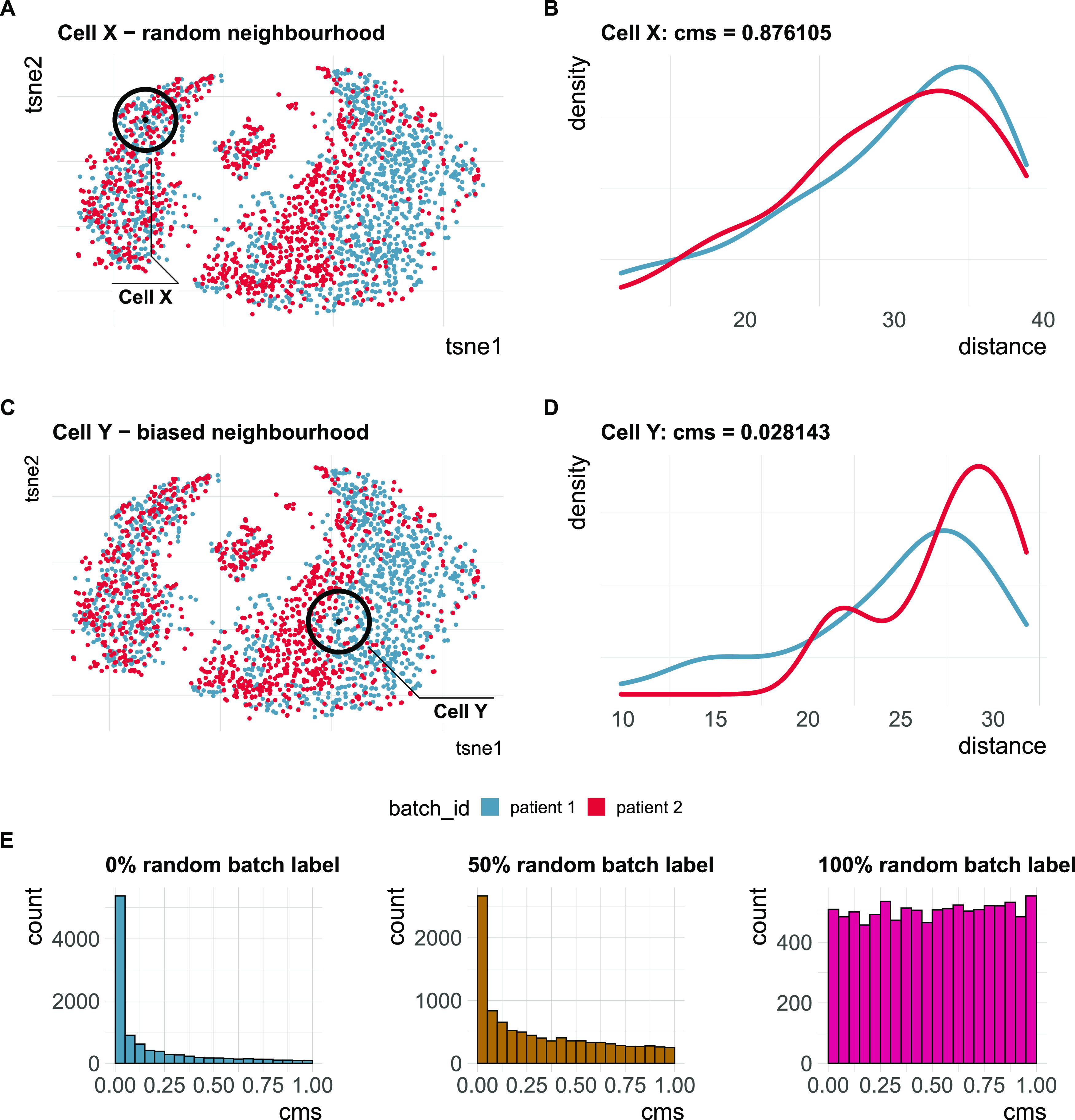
References
Publication types
MeSH terms
Associated data
LinkOut - more resources
Full Text Sources
Other Literature Sources