

Serine biosynthesis defect due to haploinsufficiency of PHGDH causes retinal disease
- PMID: 33758422
- PMCID: PMC8084205
- DOI: 10.1038/s42255-021-00361-3
Serine biosynthesis defect due to haploinsufficiency of PHGDH causes retinal disease
Abstract
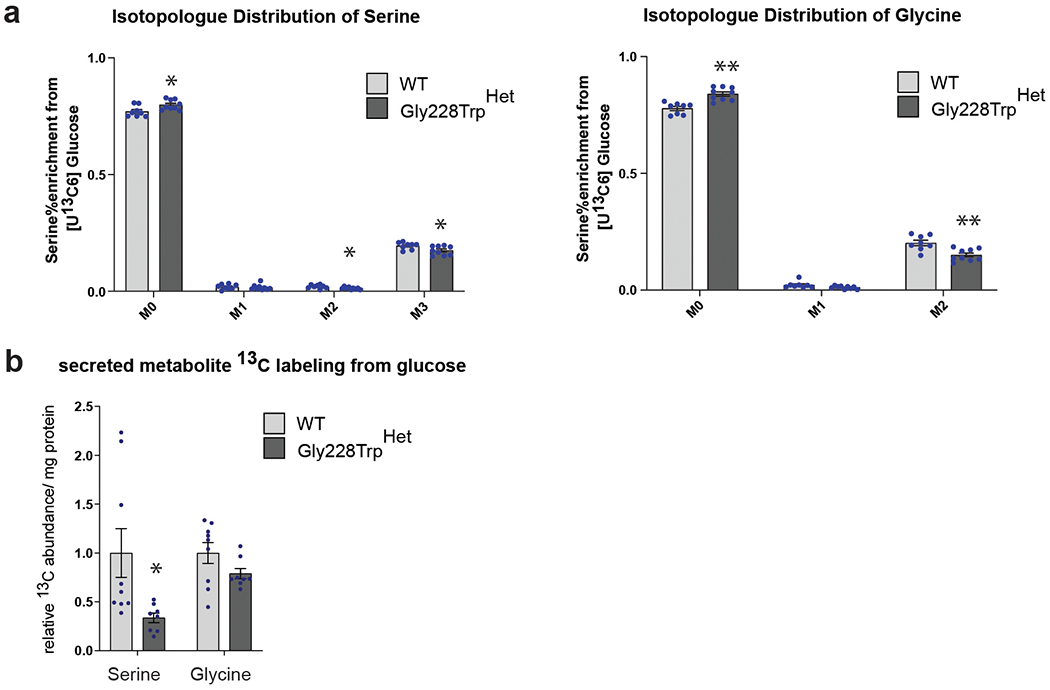
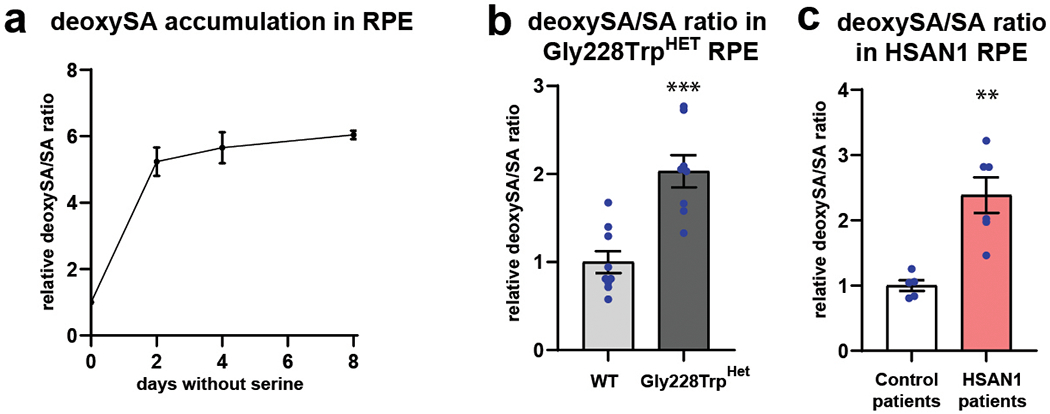
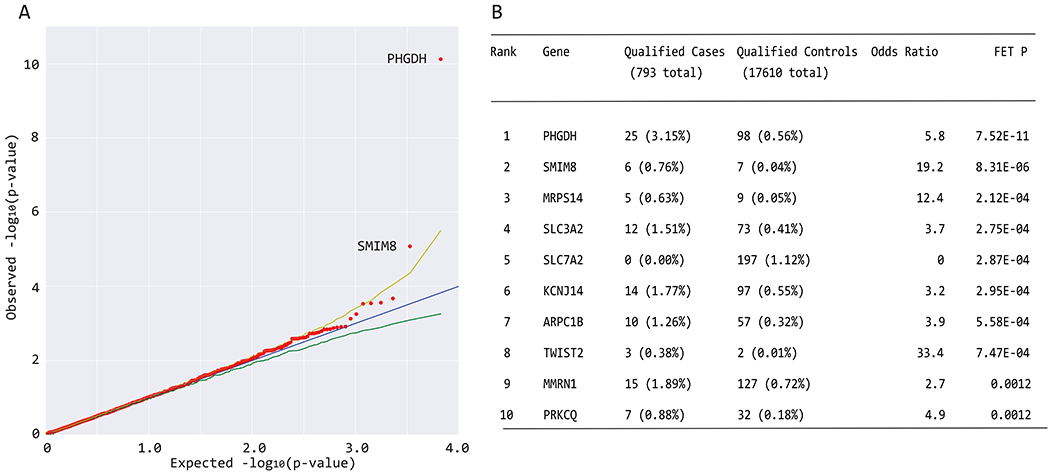
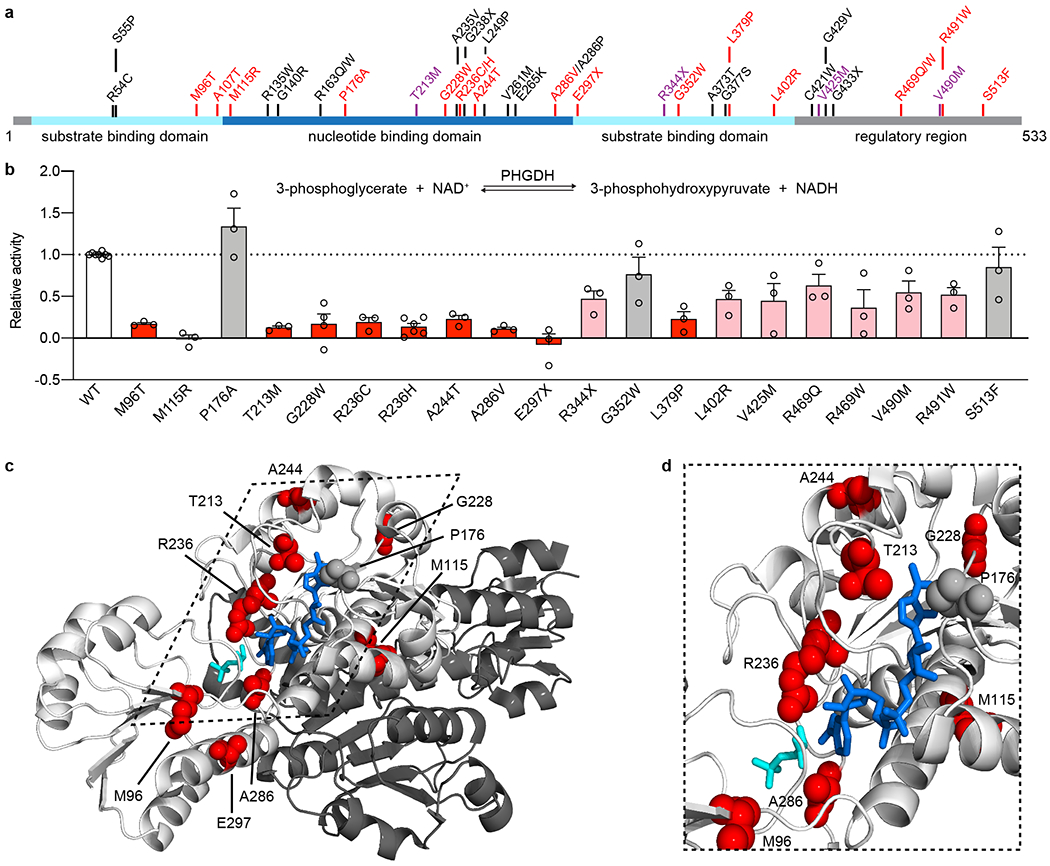
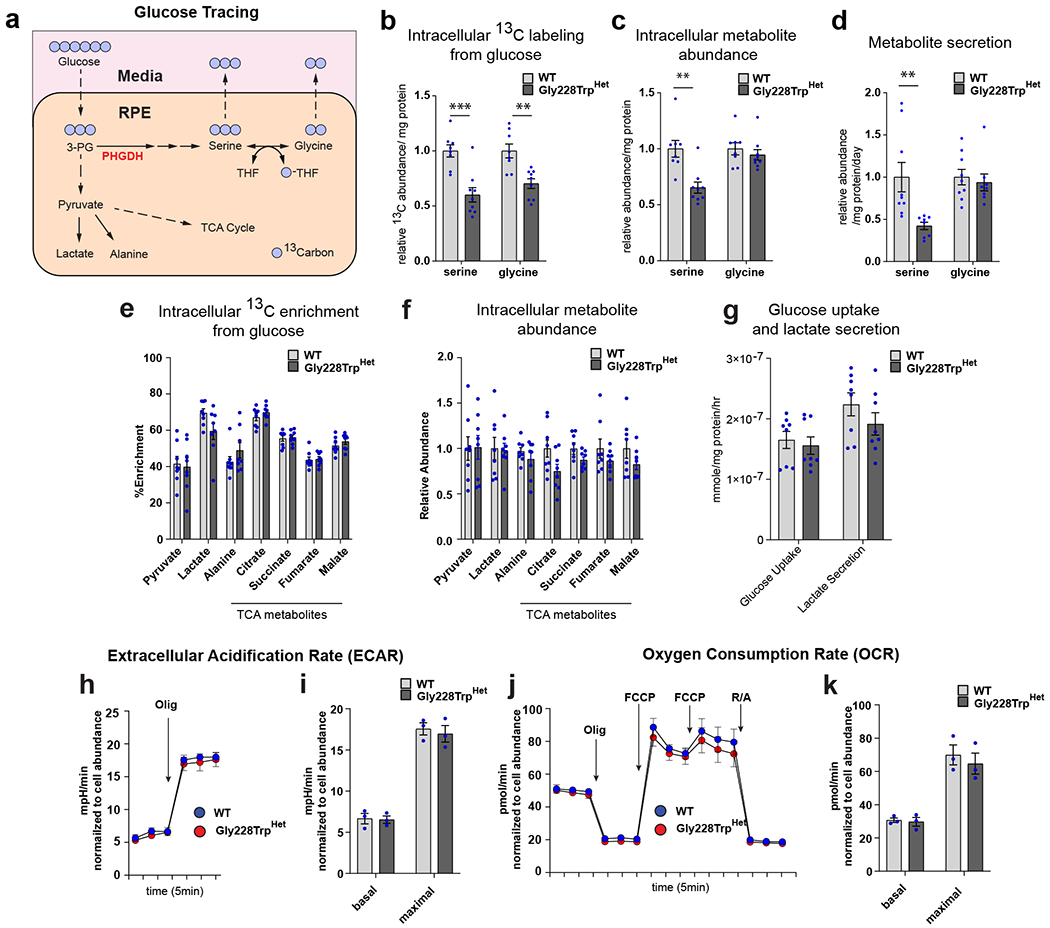
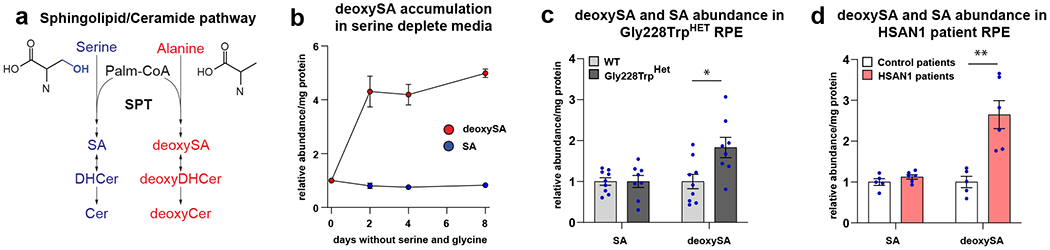
Macular telangiectasia type 2 (MacTel) is a progressive, late-onset retinal degenerative disease linked to decreased serum levels of serine that elevate circulating levels of a toxic ceramide species, deoxysphingolipids (deoxySLs); however, causal genetic variants that reduce serine levels in patients have not been identified. Here we identify rare, functional variants in the gene encoding the rate-limiting serine biosynthetic enzyme, phosphoglycerate dehydrogenase (PHGDH), as the single locus accounting for a significant fraction of MacTel. Under a dominant collapsing analysis model of a genome-wide enrichment analysis of rare variants predicted to impact protein function in 793 MacTel cases and 17,610 matched controls, the PHGDH gene achieves genome-wide significance (P = 1.2 × 10-13) with variants explaining ~3.2% of affected individuals. We further show that the resulting functional defects in PHGDH cause decreased serine biosynthesis and accumulation of deoxySLs in retinal pigmented epithelial cells. PHGDH is a significant locus for MacTel that explains the typical disease phenotype and suggests a number of potential treatment options.
Conflict of interest statement
Figures
Similar articles
-
Genetic disruption of serine biosynthesis is a key driver of macular telangiectasia type 2 aetiology and progression.Genome Med. 2021 Mar 9;13(1):39. doi: 10.1186/s13073-021-00848-4. Genome Med. 2021. PMID: 33750426 Free PMC article.
-
Systemic lipid dysregulation is a risk factor for macular neurodegenerative disease.Sci Rep. 2020 Jul 22;10(1):12165. doi: 10.1038/s41598-020-69164-y. Sci Rep. 2020. PMID: 32699277 Free PMC article.
-
iPSC-derived retinal pigmented epithelial cells from patients with macular telangiectasia show decreased mitochondrial function.J Clin Invest. 2023 May 1;133(9):e163771. doi: 10.1172/JCI163771. J Clin Invest. 2023. PMID: 37115691 Free PMC article.
-
The Role of D-3-Phosphoglycerate Dehydrogenase in Cancer.Int J Biol Sci. 2020 Mar 5;16(9):1495-1506. doi: 10.7150/ijbs.41051. eCollection 2020. Int J Biol Sci. 2020. PMID: 32226297 Free PMC article. Review.
-
Mild phenotypes of phosphoglycerate dehydrogenase deficiency by a novel mutation of PHGDH gene: Case report and literature review.Int J Dev Neurosci. 2023 Feb;83(1):44-52. doi: 10.1002/jdn.10236. Epub 2022 Nov 9. Int J Dev Neurosci. 2023. PMID: 36308023 Review.
Cited by
-
Dynamic lipid turnover in photoreceptors and retinal pigment epithelium throughout life.Prog Retin Eye Res. 2022 Jul;89:101037. doi: 10.1016/j.preteyeres.2021.101037. Epub 2021 Dec 29. Prog Retin Eye Res. 2022. PMID: 34971765 Free PMC article. Review.
-
High-throughput ultrastructural analysis of macular telangiectasia type 2.Front Ophthalmol (Lausanne). 2024 Jul 30;4:1428777. doi: 10.3389/fopht.2024.1428777. eCollection 2024. Front Ophthalmol (Lausanne). 2024. PMID: 39140090 Free PMC article.
-
Cell-specific cis-regulatory elements and mechanisms of non-coding genetic disease in human retina and retinal organoids.Dev Cell. 2022 Mar 28;57(6):820-836.e6. doi: 10.1016/j.devcel.2022.02.018. Epub 2022 Mar 17. Dev Cell. 2022. PMID: 35303433 Free PMC article.
-
Exome analysis links kidney malformations to developmental disorders and reveals causal genes.Nat Commun. 2025 Aug 7;16(1):7290. doi: 10.1038/s41467-025-62319-3. Nat Commun. 2025. PMID: 40774958 Free PMC article.
-
Animal Models of Retinopathy of Prematurity: Advances and Metabolic Regulators.Biomedicines. 2024 Aug 23;12(9):1937. doi: 10.3390/biomedicines12091937. Biomedicines. 2024. PMID: 39335451 Free PMC article. Review.
References
-
- Aung KZ, Wickremasinghe SS, Makeyeva G, Robman L & Guymer RH The prevalence estimates of macular telangiectasia type 2: the Melbourne Collaborative Cohort Study. Retina 30, 473–8 (2010). - PubMed
-
- Scerri TS et al. Genome-wide analyses identify common variants associated with macular telangiectasia type 2. Nat Genet 49, 559–567 (2017). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
