

This is a preprint.
High-resolution epigenome analysis in nasal samples derived from children with respiratory viral infections reveals striking changes upon SARS-CoV-2 infection
- PMID: 33758880
- PMCID: PMC7987039
- DOI: 10.1101/2021.03.09.21253155
High-resolution epigenome analysis in nasal samples derived from children with respiratory viral infections reveals striking changes upon SARS-CoV-2 infection
Abstract
Background: DNA methylation patterns of the human genome can be modified by environmental stimuli and provide dense information on gene regulatory circuitries. We studied genome-wide DNA methylation in nasal samples from infants (<6 months) applying whole-genome bisulfite sequencing (WGBS) to characterize epigenome response to 10 different respiratory viral infections including SARS-CoV-2.
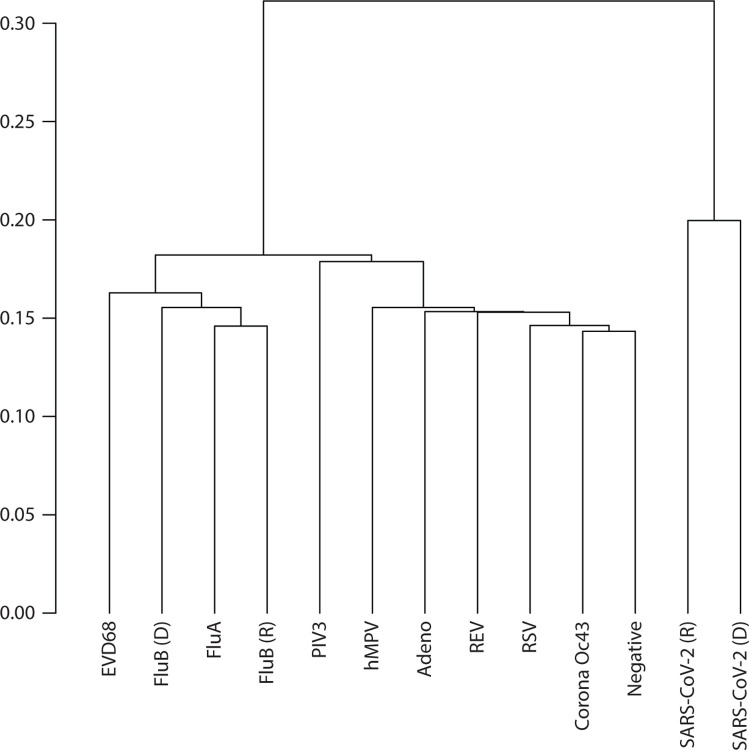
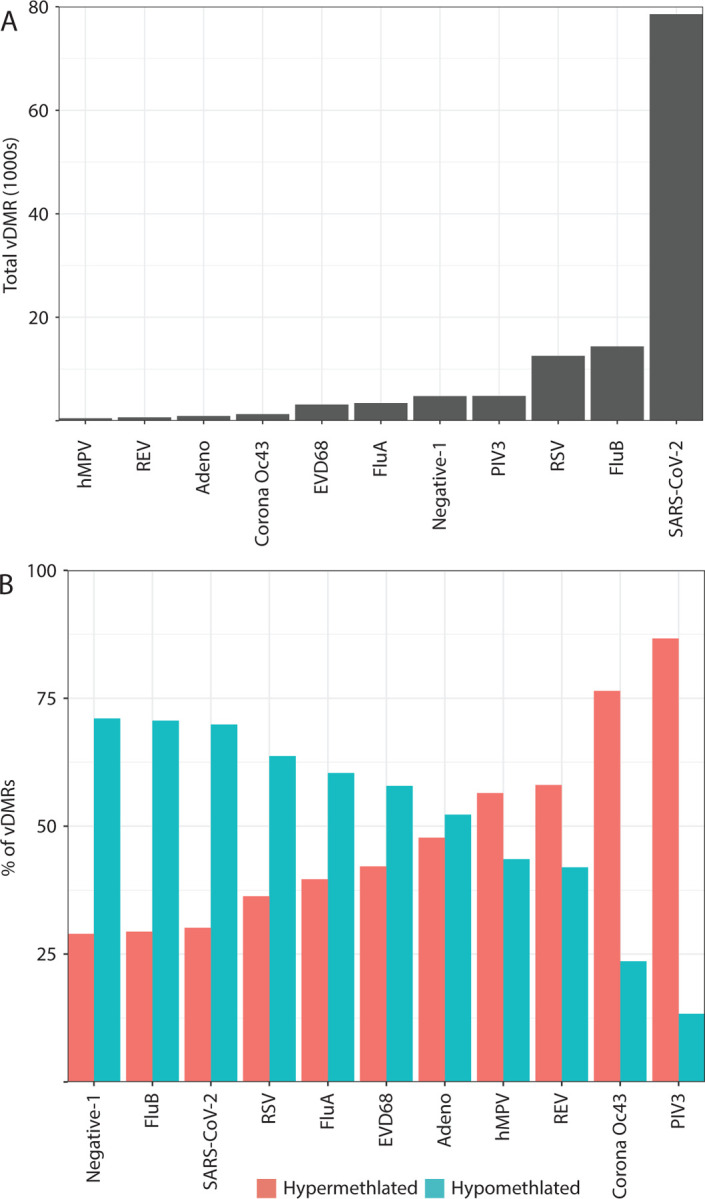
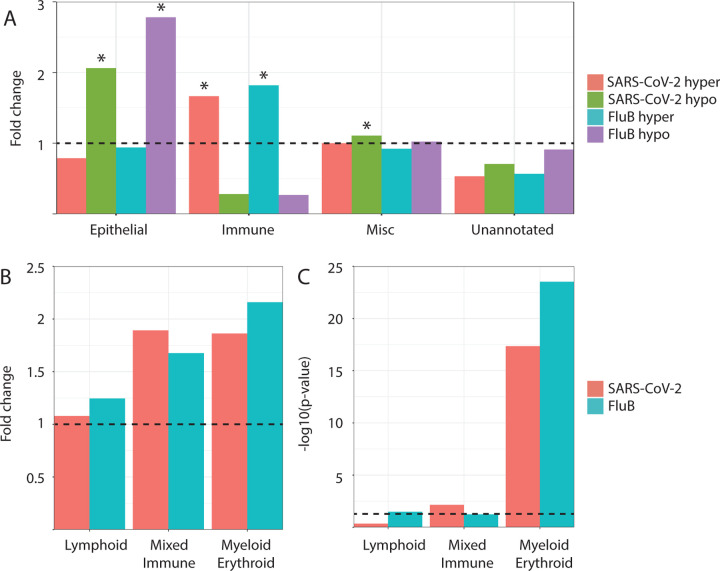
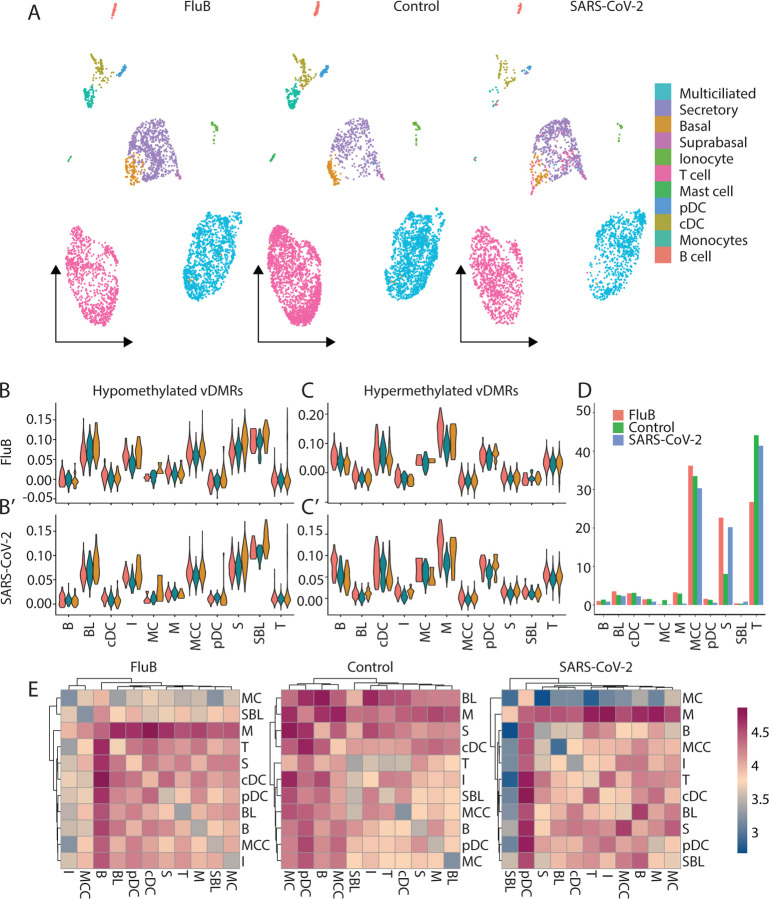
Results: We identified virus-specific differentially methylated regions (vDMR) with human metapneumovirus (hMPV) and SARS-CoV-2 followed by Influenza B (Flu B) causing the weakest vs. strongest epigenome response with 496 vs. 78541 and 14361 vDMR, respectively. We found a strong replication rate of FluB (52%) and SARS-CoV-2 (42%) vDMR in independent samples indicating robust epigenome perturbation upon infection. Among the FluB and SARS-CoV-2 vDMRs, around 70% were hypomethylated and significantly enriched among epithelial cell-specific regulatory elements whereas the hypermethylated vDMRs for these viruses mapped more frequently to immune cell regulatory elements, especially those of the myeloid lineage. The hypermethylated vDMRs were also enriched among genes and genetic loci in monocyte activation pathways and monocyte count. Finally, we perform single-cell RNA-sequencing characterization of nasal mucosa in response to these two viruses to functionally analyze the epigenome perturbations. Which supports the trends we identified in methylation data and highlights and important role for monocytes.
Conclusions: All together, we find evidence indicating genetic predisposition to innate immune response upon a respiratory viral infection. Our genome-wide monitoring of infant viral response provides first catalogue of associated host regulatory elements. Assessing epigenetic variation in individual patients may reveal evidence for viral triggers of childhood disease.
Keywords: DNA methylation; GWAS; SARS-CoV-2; infant; influenza; viral infection.
Conflict of interest statement
Competing interests The authors declare that they have no competing interests
Figures
References
-
- Broad Institute. 2019. Picard toolkit. http://broadinstitute.github.io/picard/.
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous