

Genome-resolved metagenomics using environmental and clinical samples
- PMID: 33758906
- PMCID: PMC8425419
- DOI: 10.1093/bib/bbab030
Genome-resolved metagenomics using environmental and clinical samples
Abstract
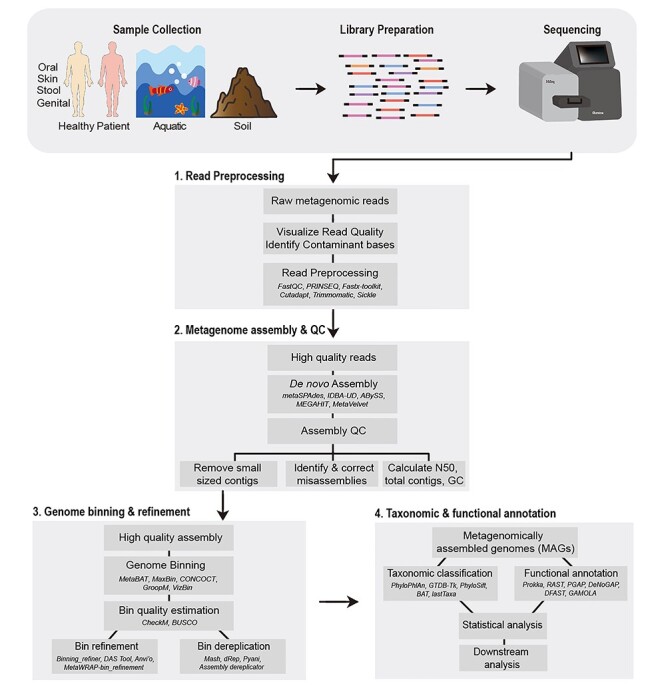
Recent advances in high-throughput sequencing technologies and computational methods have added a new dimension to metagenomic data analysis i.e. genome-resolved metagenomics. In general terms, it refers to the recovery of draft or high-quality microbial genomes and their taxonomic classification and functional annotation. In recent years, several studies have utilized the genome-resolved metagenome analysis approach and identified previously unknown microbial species from human and environmental metagenomes. In this review, we describe genome-resolved metagenome analysis as a series of four necessary steps: (i) preprocessing of the sequencing reads, (ii) de novo metagenome assembly, (iii) genome binning and (iv) taxonomic and functional analysis of the recovered genomes. For each of these four steps, we discuss the most commonly used tools and the currently available pipelines to guide the scientific community in the recovery and subsequent analyses of genomes from any metagenome sample. Furthermore, we also discuss the tools required for validation of assembly quality as well as for improving quality of the recovered genomes. We also highlight the currently available pipelines that can be used to automate the whole analysis without having advanced bioinformatics knowledge. Finally, we will highlight the most widely adapted and actively maintained tools and pipelines that can be helpful to the scientific community in decision making before they commence the analysis.
Keywords: MAG annotation; MAG refinement; MAG taxonomic classification; de novo assembly; genome binning; metagenome assembly validation; metagenome-assembled genomes; read preprocessing.
© The Author(s) 2021. Published by Oxford University Press. All rights reserved.
Figures
Similar articles
-
Evaluating de Novo Assembly and Binning Strategies for Time Series Drinking Water Metagenomes.Microbiol Spectr. 2021 Dec 22;9(3):e0143421. doi: 10.1128/Spectrum.01434-21. Epub 2021 Nov 3. Microbiol Spectr. 2021. PMID: 34730411 Free PMC article.
-
MAGNETO: An Automated Workflow for Genome-Resolved Metagenomics.mSystems. 2022 Aug 30;7(4):e0043222. doi: 10.1128/msystems.00432-22. Epub 2022 Jun 15. mSystems. 2022. PMID: 35703559 Free PMC article.
-
Genome-resolved metagenomics from short-read sequencing data in the era of artificial intelligence.Funct Integr Genomics. 2025 Jun 10;25(1):124. doi: 10.1007/s10142-025-01625-x. Funct Integr Genomics. 2025. PMID: 40493087 Review.
-
MetaCluster-TA: taxonomic annotation for metagenomic data based on assembly-assisted binning.BMC Genomics. 2014;15 Suppl 1(Suppl 1):S12. doi: 10.1186/1471-2164-15-S1-S12. Epub 2014 Jan 24. BMC Genomics. 2014. PMID: 24564377 Free PMC article.
-
Recovering metagenome-assembled genomes from shotgun metagenomic sequencing data: Methods, applications, challenges, and opportunities.Microbiol Res. 2022 Jul;260:127023. doi: 10.1016/j.micres.2022.127023. Epub 2022 Apr 8. Microbiol Res. 2022. PMID: 35430490 Review.
Cited by
-
Constructing metagenome-assembled genomes for almost all components in a real bacterial consortium for binning benchmarking.BMC Genomics. 2022 Nov 10;23(1):746. doi: 10.1186/s12864-022-08967-x. BMC Genomics. 2022. PMID: 36352370 Free PMC article.
-
Efficient De Novo Assembly and Recovery of Microbial Genomes from Complex Metagenomes Using a Reduced Set of k-mers.Interdiscip Sci. 2025 Jun 2. doi: 10.1007/s12539-025-00722-6. Online ahead of print. Interdiscip Sci. 2025. PMID: 40455399
-
Contamination detection and microbiome exploration with GRIMER.Gigascience. 2022 Dec 28;12:giad017. doi: 10.1093/gigascience/giad017. Epub 2023 Mar 30. Gigascience. 2022. PMID: 36994872 Free PMC article.
-
Next Generation Sequencing Approaches to Characterize the Respiratory Tract Virome.Microorganisms. 2022 Nov 24;10(12):2327. doi: 10.3390/microorganisms10122327. Microorganisms. 2022. PMID: 36557580 Free PMC article. Review.
-
MGnify Genomes: A Resource for Biome-specific Microbial Genome Catalogues.J Mol Biol. 2023 Jul 15;435(14):168016. doi: 10.1016/j.jmb.2023.168016. Epub 2023 Feb 16. J Mol Biol. 2023. PMID: 36806692 Free PMC article.
References
-
- Hugenholtz P, Tyson GW. Metagenomics. Nature 2008;455:481–3. - PubMed
-
- Sunagawa S, Coelho LP, Chaffron S, et al. . Structure and function of the global ocean microbiome. Science 2015;348. - PubMed
-
- Hu Y, Yang X, Qin J, et al. . Metagenome-wide analysis of antibiotic resistance genes in a large cohort of human gut microbiota. Nat Commun 2013;4:1–7. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
