

Machine learning-based reclassification of germline variants of unknown significance: The RENOVO algorithm
- PMID: 33761318
- PMCID: PMC8059374
- DOI: 10.1016/j.ajhg.2021.03.010
Machine learning-based reclassification of germline variants of unknown significance: The RENOVO algorithm
Abstract
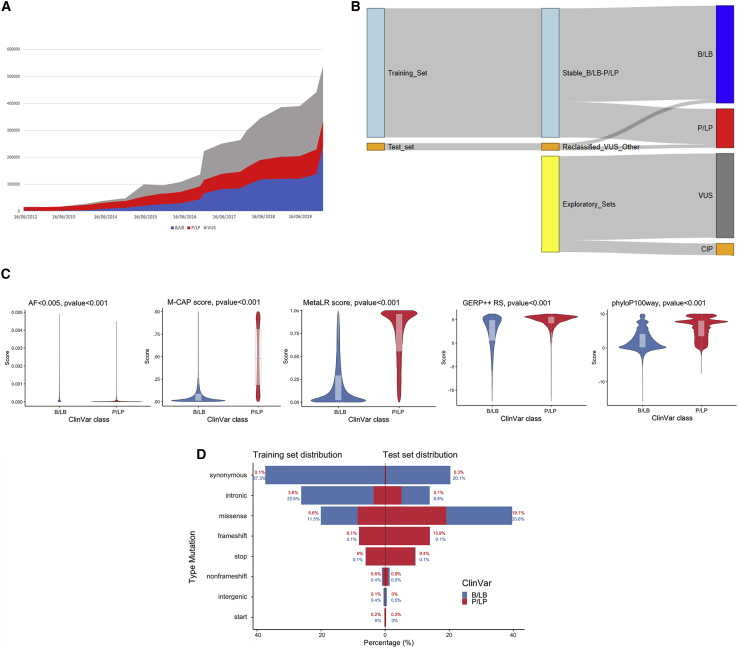
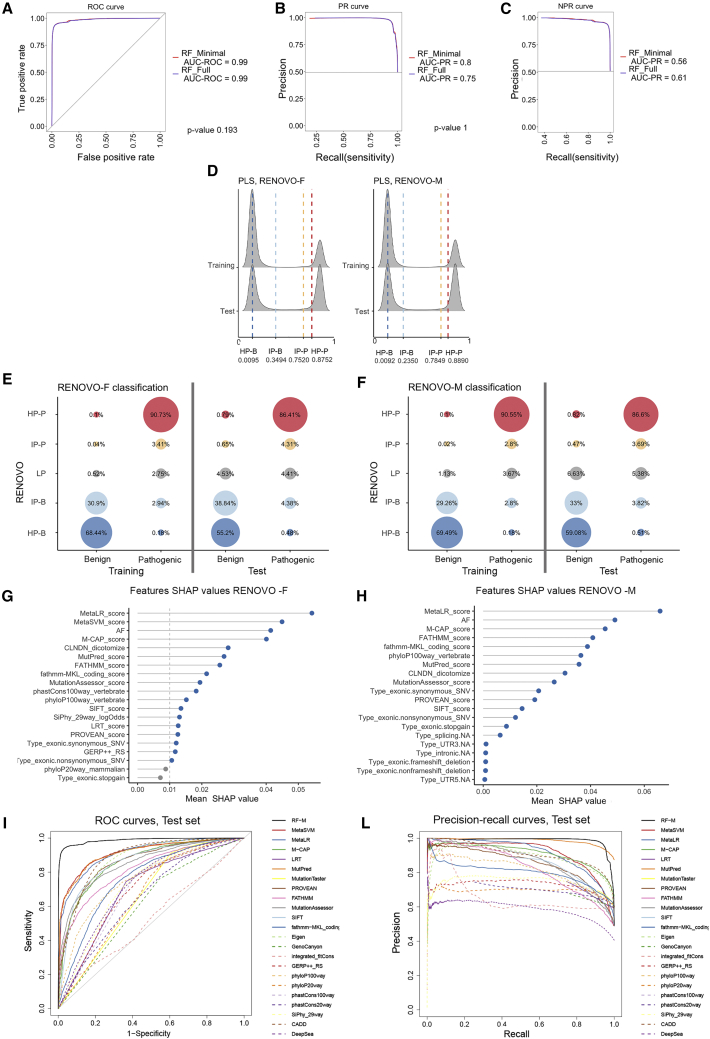
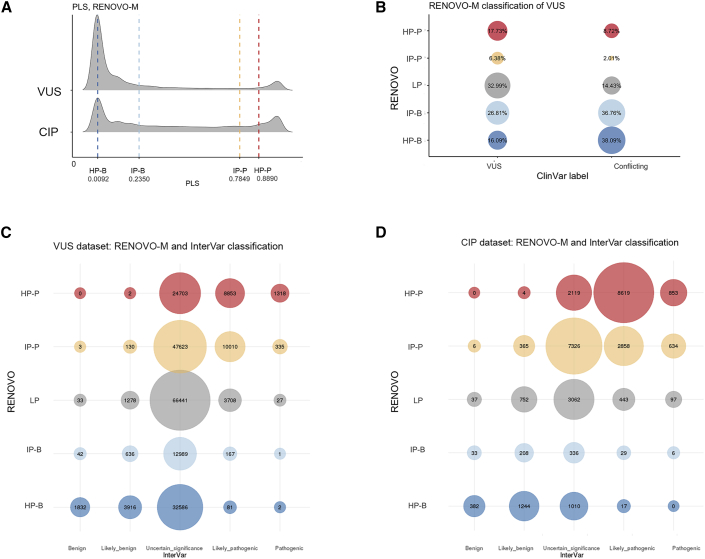
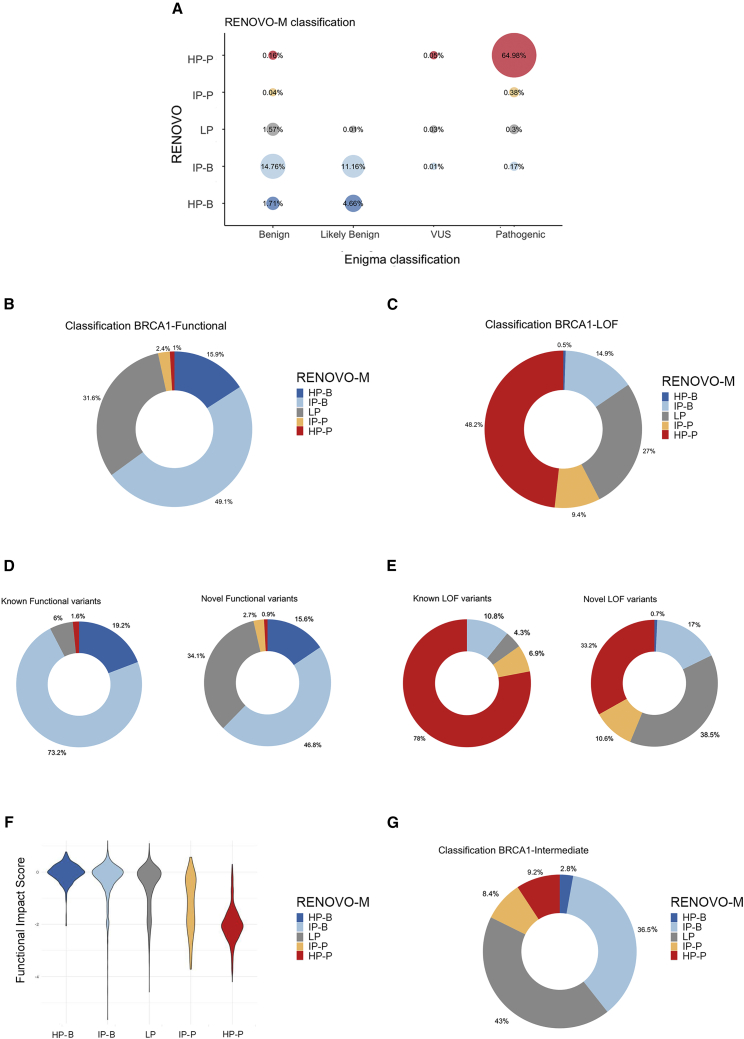
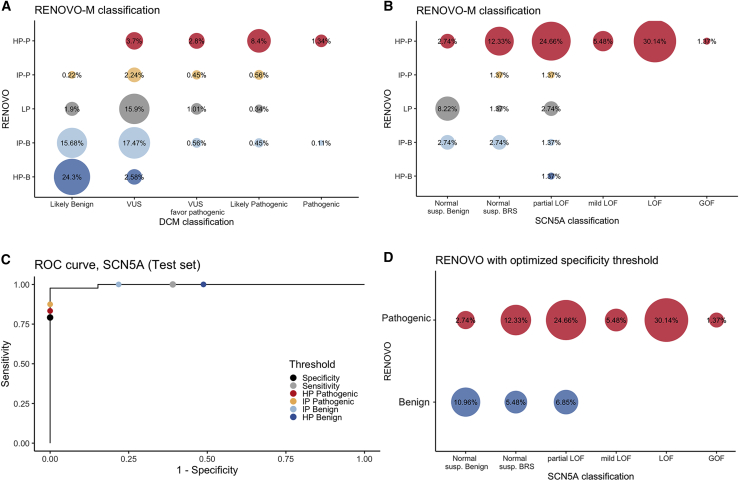
The increasing scope of genetic testing allowed by next-generation sequencing (NGS) dramatically increased the number of genetic variants to be interpreted as pathogenic or benign for adequate patient management. Still, the interpretation process often fails to deliver a clear classification, resulting in either variants of unknown significance (VUSs) or variants with conflicting interpretation of pathogenicity (CIP); these represent a major clinical problem because they do not provide useful information for decision-making, causing a large fraction of genetically determined disease to remain undertreated. We developed a machine learning (random forest)-based tool, RENOVO, that classifies variants as pathogenic or benign on the basis of publicly available information and provides a pathogenicity likelihood score (PLS). Using the same feature classes recommended by guidelines, we trained RENOVO on established pathogenic/benign variants in ClinVar (training set accuracy = 99%) and tested its performance on variants whose interpretation has changed over time (test set accuracy = 95%). We further validated the algorithm on additional datasets including unreported variants validated either through expert consensus (ENIGMA) or laboratory-based functional techniques (on BRCA1/2 and SCN5A). On all datasets, RENOVO outperformed existing automated interpretation tools. On the basis of the above validation metrics, we assigned a defined PLS to all existing ClinVar VUSs, proposing a reclassification for 67% with >90% estimated precision. RENOVO provides a validated tool to reduce the fraction of uninterpreted or misinterpreted variants, tackling an area of unmet need in modern clinical genetics.
Keywords: ClinVar; VUS; machine learning; reclassification; variant interpretation.
Copyright © 2021 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Tavtigian S.V., Greenblatt M.S., Harrison S.M., Nussbaum R.L., Prabhu S.A., Boucher K.M., Biesecker L.G., ClinGen Sequence Variant Interpretation Working Group (ClinGen SVI) Modeling the ACMG/AMP variant classification guidelines as a Bayesian classification framework. Genet. Med. 2018;20:1054–1060. - PMC - PubMed
-
- Richards S., Aziz N., Bale S., Bick D., Das S., Gastier-Foster J., Grody W.W., Hegde M., Lyon E., Spector E., ACMG Laboratory Quality Assurance Committee Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015;17:405–424. - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
