

Tunable Methacrylamides for Covalent Ligand Directed Release Chemistry
- PMID: 33761747
- PMCID: PMC8041284
- DOI: 10.1021/jacs.0c10644
Tunable Methacrylamides for Covalent Ligand Directed Release Chemistry
Abstract
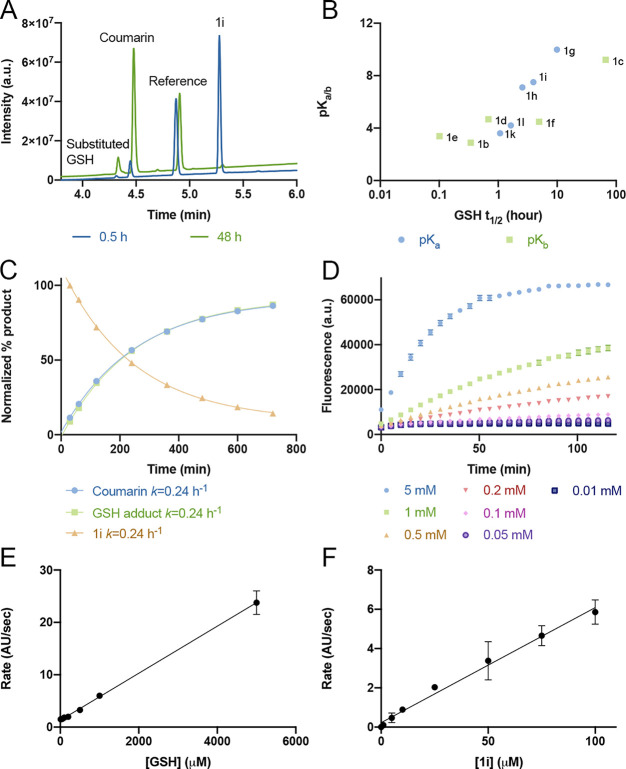
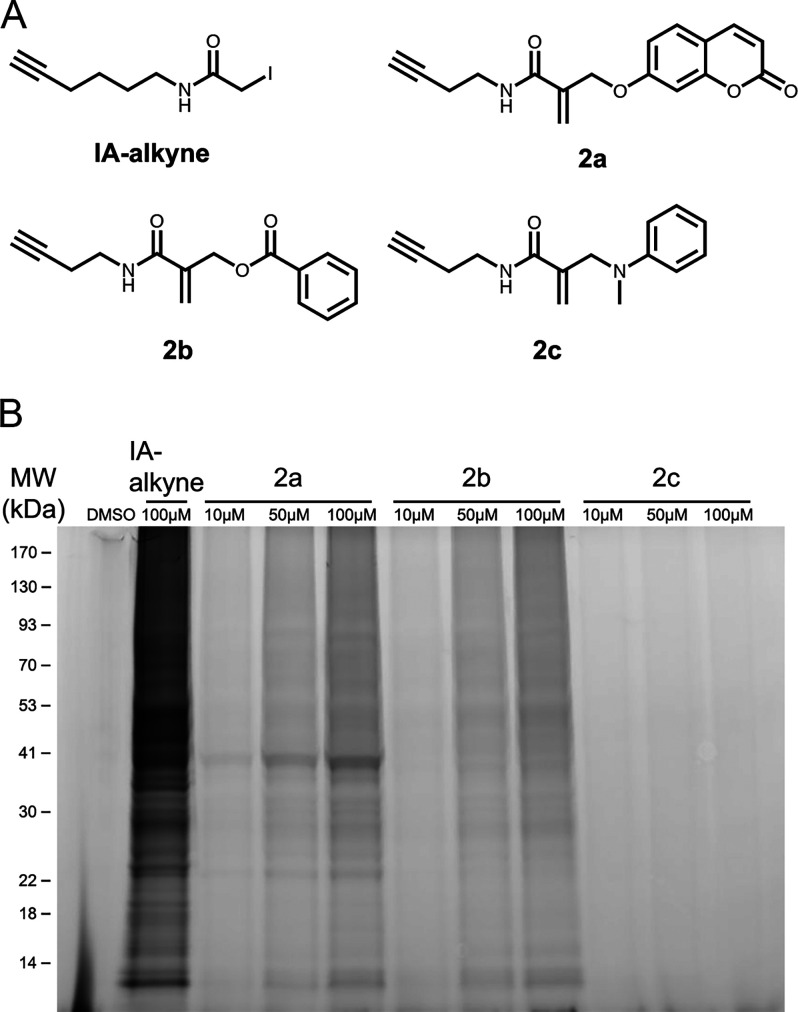
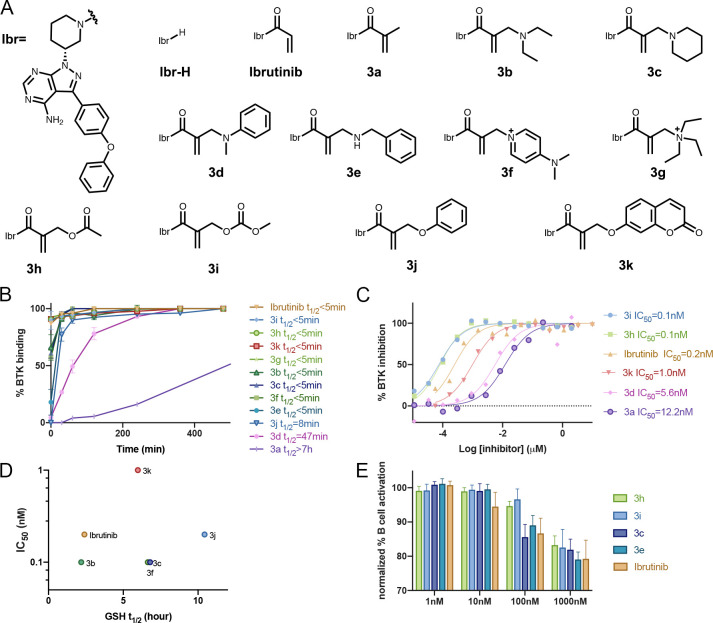
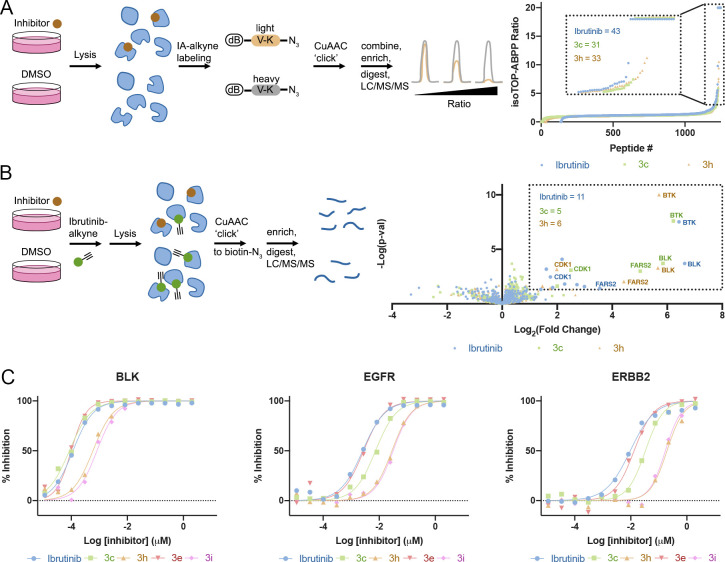
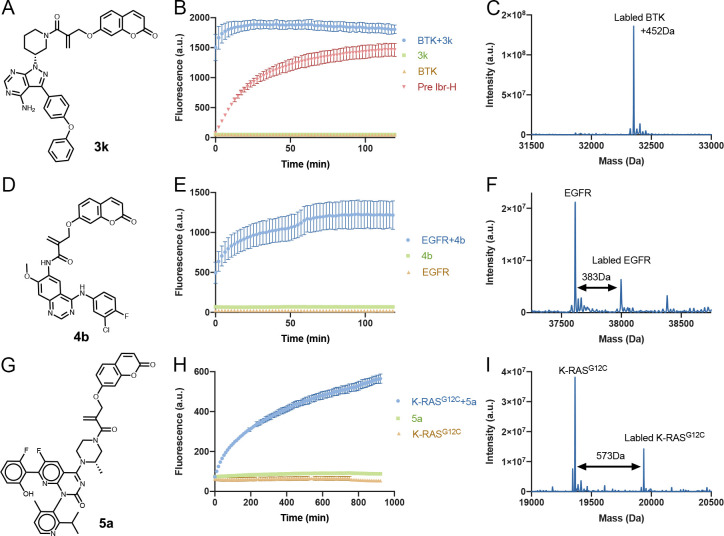
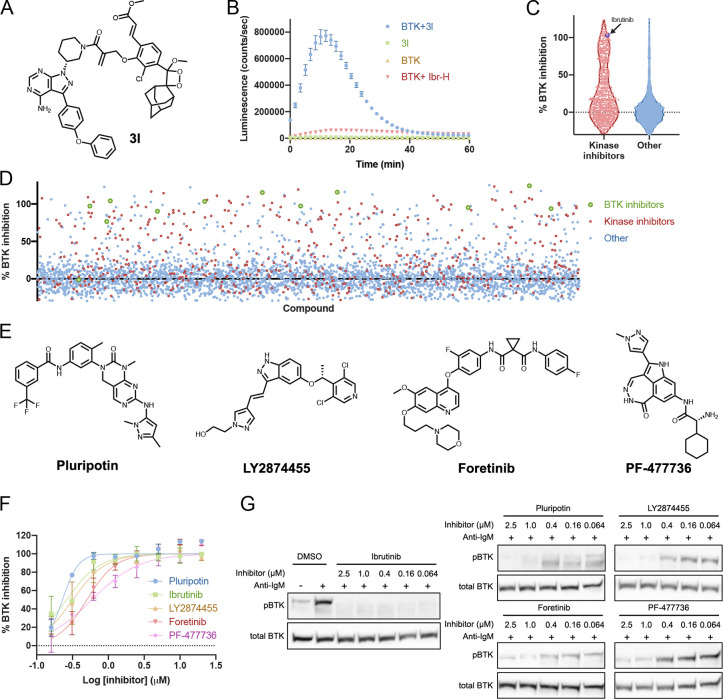
Targeted covalent inhibitors are an important class of drugs and chemical probes. However, relatively few electrophiles meet the criteria for successful covalent inhibitor design. Here we describe α-substituted methacrylamides as a new class of electrophiles suitable for targeted covalent inhibitors. While typically α-substitutions inactivate acrylamides, we show that hetero α-substituted methacrylamides have higher thiol reactivity and undergo a conjugated addition-elimination reaction ultimately releasing the substituent. Their reactivity toward thiols is tunable and correlates with the pKa/pKb of the leaving group. In the context of the BTK inhibitor ibrutinib, these electrophiles showed lower intrinsic thiol reactivity than the unsubstituted ibrutinib acrylamide. This translated to comparable potency in protein labeling, in vitro kinase assays, and functional cellular assays, with improved selectivity. The conjugate addition-elimination reaction upon covalent binding to their target cysteine allows functionalizing α-substituted methacrylamides as turn-on probes. To demonstrate this, we prepared covalent ligand directed release (CoLDR) turn-on fluorescent probes for BTK, EGFR, and K-RasG12C. We further demonstrate a BTK CoLDR chemiluminescent probe that enabled a high-throughput screen for BTK inhibitors. Altogether we show that α-substituted methacrylamides represent a new and versatile addition to the toolbox of targeted covalent inhibitor design.
Conflict of interest statement
The authors declare the following competing financial interest(s): R.N.R., E.R., A.R., and N.L. are inventors on a patent application describing CoLDR chemistry.
Figures

References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
