

Observation of the natural course of type 3 spinal muscular atrophy: data from the polish registry of spinal muscular atrophy
- PMID: 33761963
- PMCID: PMC7992780
- DOI: 10.1186/s13023-021-01771-y
Observation of the natural course of type 3 spinal muscular atrophy: data from the polish registry of spinal muscular atrophy
Abstract
Background: Spinal muscular atrophy (SMA) is one of the most frequent and severe genetic diseases leading to premature death or severe motor disability. New therapies have been developed in recent years that change the natural history of the disease. The aim of this study is to describe patients included in the Polish Registry of SMA, with a focus on the course of type 3 SMA (SMA3) before the availability of disease-modifying treatments.
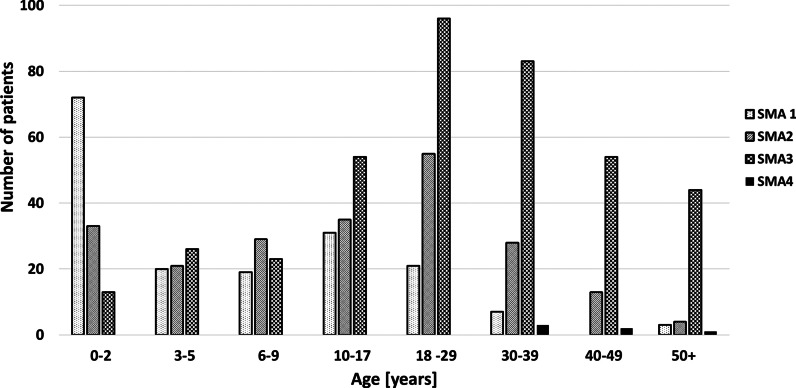
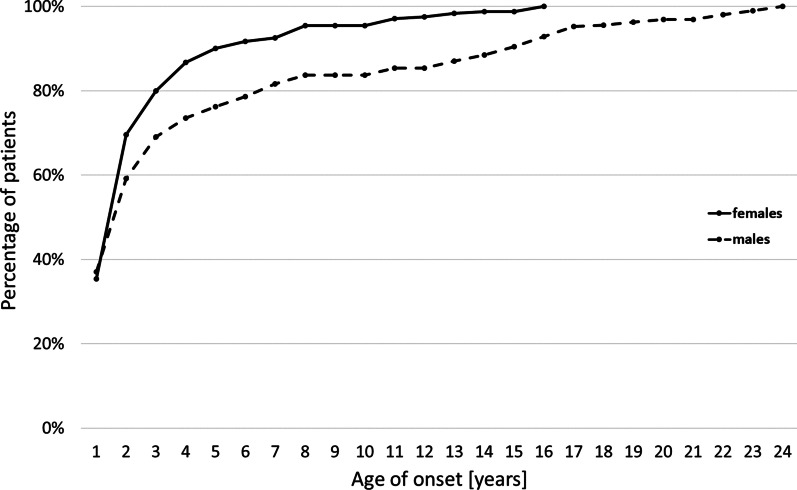
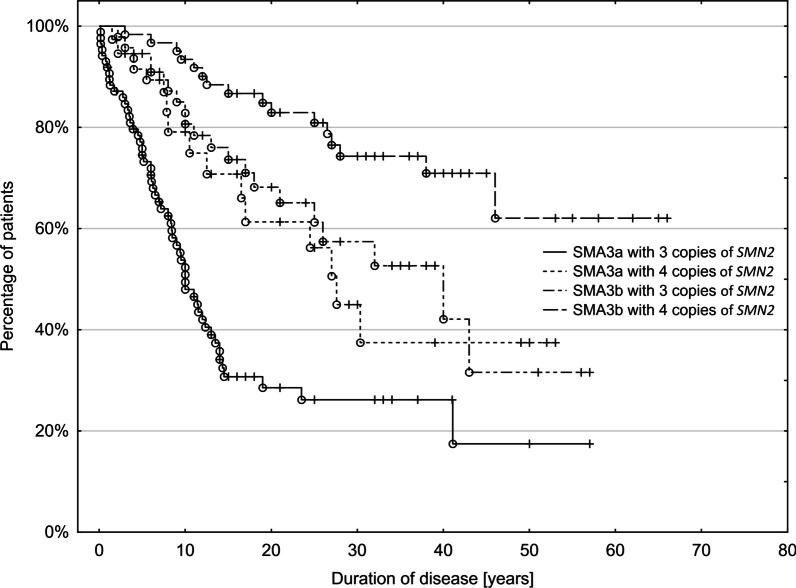
Results: 790 patients with SMA were included in the registry (173 with type 1 [SMA1], 218 with type 2 [SMA2], 393 with SMA3, and six with type 4 SMA [SMA4]), most (52%) of whom were adults. Data on SMN2 gene copy number were available for 672 (85%) patients. The mean age of onset was 5 months for SMA1, 11.5 months for SMA2, and 4.5 years for SMA3. In patients with SMA3, the first symptoms occurred earlier in those with three copies of SMN2 than in those with four copies of SMN2 (3.2 years vs. 6.7 years). The age of onset of SMA3 was younger in girls than in boys (3.1 years vs. 5.7 years), with no new cases observed in women older than 16 years. Male patients outnumbered female patients, especially among patients with SMA3b (49 female vs. 85 male patients) and among patients with SMA3 with four copies of SMN2 (30 female vs. 69 male patients). 44% of patients with SMA3 were still able to walk; in those who were not still able to walk, the mean age of immobilization was 14.0 years. Patients with SMA3a (age of onset < 3 years) and three copies of SMN2 had significantly worse prognosis for remaining ambulant than patients with SMA3b (age of onset ≥ 3 years) and four copies of SMN2.
Conclusions: The Registry of SMA is an effective tool for assessing the disease course in the real world setting. SMN2 copy number is an important prognostic factor for the age of onset and ambulation in SMA3. Sex and age of disease onset also strongly affect the course of SMA. Data supplied by this study can aid treatment decisions.
Keywords: Neuromuscular disease; Registry; SMN2 copy number; Spinal muscular atrophy (SMA); TREAT-NMD; Type 3 SMA.
Conflict of interest statement
Anna Lusakowska is a sub-investigator in clinical trials for patients with SMA (funded by Roche) and Duchenne muscular dystrophy (funded by PTC), receives institutional Grant support from Biogen, and is a lecturer on neuromuscular diseases supported by Biogen and Roche. Maria Jedrzejowska reports participation in scientific advisory boards for Biogen and Avexis/Novartis, receives honoraria for lectures from Biogen and travel support from Biogen and Roche, and acts as a sub-investigator in Roche trials of olesoxime and risdiplam for SMA. Anna Kaminska declares no conflicts of interest. Katarzyna Janiszewska declares no conflicts of interest. Przemysław Grochowski declares no conflicts of interest. Janusz Zimowski declares no conflicts of interest. Janusz Sierdzinski declares no conflicts of interest. Anna Kostera Pruszczyk reports lecture honoraria and travel support from Biogen, Roche, PTC, Avexis, and Novartis, institutional Grant support from Biogen, and advisory board honoraria from Biogen, Roche, Avexis, and Novartis, and is a principal investigator for Roche SMA studies.
Figures
References
-
- Zerres K, Rudnik-Schöneborn S, Forrest E, Lusakowska A, Borkowska J, Hausmanowa-Petrusewicz I. A collaborative study on the natural history of childhood and juvenile onset proximal spinal muscular atrophy (type II and III): 569 patients. J Neurol Sci. 1997;146(1):67–72. doi: 10.1016/S0022-510X(96)00284-5. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
