

Neuron-Oligodendrocyte Interactions in the Structure and Integrity of Axons
- PMID: 33763430
- PMCID: PMC7982542
- DOI: 10.3389/fcell.2021.653101
Neuron-Oligodendrocyte Interactions in the Structure and Integrity of Axons
Abstract
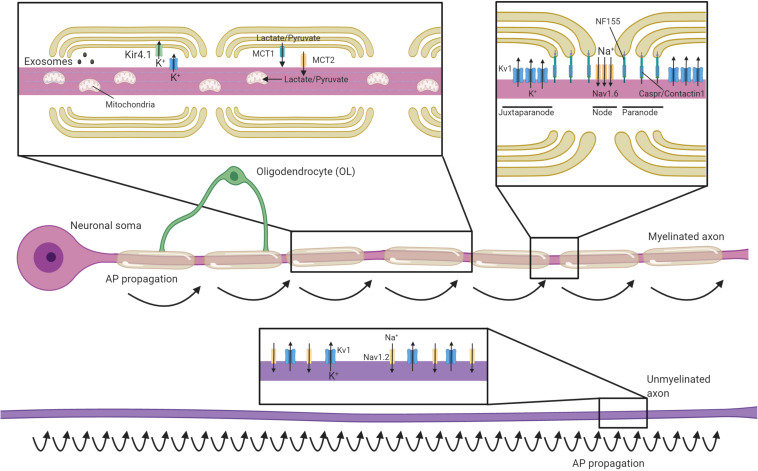
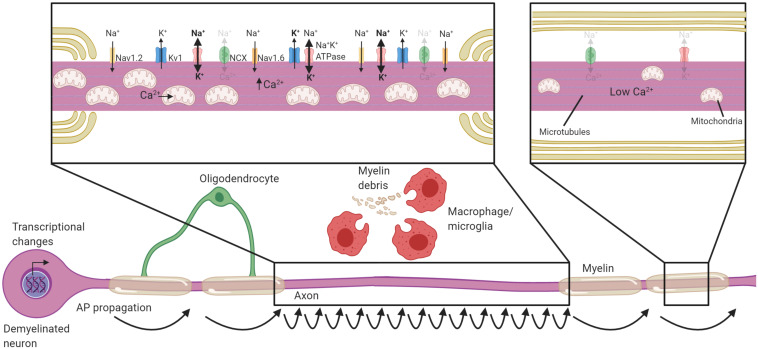
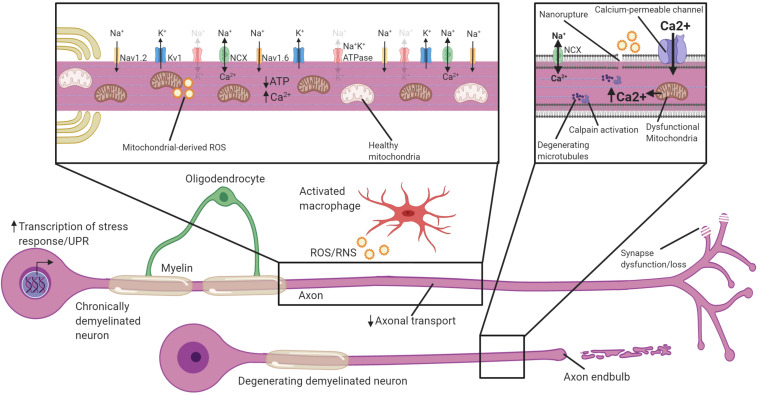
The myelination of axons by oligodendrocytes is a highly complex cell-to-cell interaction. Oligodendrocytes and axons have a reciprocal signaling relationship in which oligodendrocytes receive cues from axons that direct their myelination, and oligodendrocytes subsequently shape axonal structure and conduction. Oligodendrocytes are necessary for the maturation of excitatory domains on the axon including nodes of Ranvier, help buffer potassium, and support neuronal energy metabolism. Disruption of the oligodendrocyte-axon unit in traumatic injuries, Alzheimer's disease and demyelinating diseases such as multiple sclerosis results in axonal dysfunction and can culminate in neurodegeneration. In this review, we discuss the mechanisms by which demyelination and loss of oligodendrocytes compromise axons. We highlight the intra-axonal cascades initiated by demyelination that can result in irreversible axonal damage. Both the restoration of oligodendrocyte myelination or neuroprotective therapies targeting these intra-axonal cascades are likely to have therapeutic potential in disorders in which oligodendrocyte support of axons is disrupted.
Keywords: Wallerian degeneration; axonal degeneration; demyelination; mitochondria; multiple sclerosis; oligodendrocyte; remyelination.
Copyright © 2021 Duncan, Simkins and Emery.
Conflict of interest statement
BE is a co-founder of Autobahn Therapeutics. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Aharoni R., Vainshtein A., Stock A., Eilam R., From R., Shinder V., et al. (2011). Distinct pathological patterns in relapsing-remitting and chronic models of experimental autoimmune enchephalomyelitis and the neuroprotective effect of glatiramer acetate. J. Autoimmun. 37 228–241. 10.1016/j.jaut.2011.06.003 - DOI - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources